Introduction
Parkinson's disease (PD) is a neurological disorder that results as dopaminergic neurons die in the basal ganglia (substantia nigra compacta and pedunculopontine nucleus) of the brain (Figure 1). The brain loses 50-70% of the dopaminergic neurons over the course of the disease [1]. Changes in the brain progress from the medulla oblongata/pontine tegmentum and olfactory bulb to the substantia nigra, midbrain and basal forebrain and finally to the neocortex [1]. These changes coincide with the distribution of Lewy Bodies, an alpha-synuclein inclusion [1]. Additionally, abnormalities in Complex 1 involving mitochondrial genetics appear to play a role in Parkinson's disease [1].

Figure 1. Comparison of the substantia nigra in a healthy individual to that in Parkinson's disease.
From: http://www.healthcentral.com/common/images/1/19515_9684_5.jpg
Diagnosed largely by clinical manifestations, it is characterized by motor and non-motor signs. Functional neuroimaging, although not for diagnosis, can be utilized for research purposes [2]. The cardinal signs are resting tremor, rigidity, bradykinesia, and postural instability. Other motor signs and symptoms include hypokinesia, rigidity, difficulty with movement initiation and termination. Non-motor symptoms are flat facial affect, alterations in sleep/wake cycles causing excessive daytime sleepiness, constipation, orthostatic hypotension, thermal dysregulation, and bladder and sexual dysfunction. One symptom that occurs prior to the motor symptoms is a decrease in smell and taste [1]. Dementia and psychosis occur later in the disease.
A small number of cases are believed to be from genetic causes, while the majority arise sporadically from oxidative stress, mitochondrial dysfunction, and programmed cell death. [3] [4]. The role of environmental exposure is still unclear [2]. At least 11 single gene mutations have been identified [1]. These can have an autosomal dominant or autosomal recessive inheritance [1]. The most common genetic mutation has been found with the LRRK2 gene [1].
In industrialized countries, the prevalence is estimated at 0.3 % and occurs most often in individuals over 60 years old [3]. Ten million people worldwide and one million in the United States are estimated to have Parkinson’s disease. There are approximately 60,000 cases diagnosed and reported each year in the United States [5]. After diagnosis, the average lifespan is 13 years [4].
Currently, there is no cure for Parkinson’s disease, but there are medical treatments available including pharmaceuticals, occupational and physical therapy, and surgical procedures. The mainstay drug is Levodopa. It reduces the motor symptoms of the disease by replacing the dopamine. It is often combined with a peripheral decarboxylase inhibitor such as carbidopa [1]. These drugs lead to serious disabling side effects such as dyskinesias, dystonia, and immobile rigidity, but may also have neuroprotective factors [1]. Occupational and physical therapy work to provide means for improving function and methods of compensation. Balance, strengthening, and stretching are also incorporated often using visual and vocal cueing [6]. Surgical procedures historically included thalamotomy and pallidotomy to reduce tremor and motor symptoms [2]. Current options are utilized as a response to long-term complications from pharmaceutical management [2]. These include deep-brain stimulation, neuronal transplantation, and destructive surgery [4]. While the risk of adverse outcomes is low, they can be serious [1]. Possibilities include brain hemorrhage, infarct, seizure, and death [2].
|
Table of Contents
|
Etiology
Idiopathic Parkinsonism
The exact cause of PD, or idiopathic parkinsonism, is unknown in 78% of cases; however, approximately 15% of those with PD have a first-degree relative with the disease, and recent evidence suggests the involvement of single gene mutations including alpha-synuclein (SNCA), LRRK 2, parkin (PRKN), DJ-1, PINK 1, and HTRA 2 genes [7],[2],[8],[1]. Many of these mutations have been linked to mitochondrial dysfunction, primarily involving impaired function of the electron transport chain, which results in increased oxidative stress and increased susceptibility of substantia nigra and frontal cortex cells to excitotoxicity, accumulation of damaged proteins, and apoptosis [8].
SNCA Gene
Mutations involving the SNCA gene have been linked to an autosomal dominant form of PD with the presence of lewy bodies, which has been shown to be a primary pathological mechanism of the disease [1]. The protein encoded by this gene, alpha-synuclein, is a component of lewy bodies. Furthermore, there is evidence that mutation of the gene and overexpression of alpha-synuclein is associated with decreased mitochondrial function, abnormal mitochondria, damaged mitochondrial DNA, increased cytochrome c release, and increased free radical production [8].
LRRK 2
Damage to the LRRK 2 gene is the most common known cause for idiopathic parkinsonism, accounting for up to 5-7% of cases with a family history [1]. The most common mutation of this gene is autosomal dominant and results in an altered kinase activity and might influence the function of the outer mitochondrial membrane [8]. Patients with LRRK 2 involvement typically experience middle to late disease onset and demonstrate classic parkinsonian symptoms [1].
Parkin and DJ-1 Genes
Involvement of parkin and DJ-1 genes typically present as autosomal recessive mutations and are associated with an earlier disease onset. However, the course of the disease in these cases is often more benign and responds well to medical interventions [1]. Products of the parkin gene are involved in mitochondrial biogenesis and mitochondrial DNA replication; thus, mutations have consequences such abnormal mitochondria, increased oxidative stress, and increased sensitivity to oxidative stress. The DJ-1 gene is primarily responsible for protecting cells from oxidative-stress-related death. Mutations in mice have demonstrated increase susceptibility to oxidative stress in embryonic cortical neurons and degeneration of dopaminergic neurons [8]. In addition, absence of the DJ-1 gene has been associated with a down-regulation of the receptor for glial cell line-derived neurotrophic factor (GDNF), which provides neurotrophic support to dopamine neurons [9].
PINK 1 Gene
The PINK 1 gene has also been correlated with an autosomal recessive form of PD that is much rarer than those involving parkin and DJ-1 genes [8],[1]. The gene encodes for a protein kinase that is associated with the mitochondrial membrane and functions to reduce mitochondrial chromosome c release, and reduce apoptosis. Mutations of the gene result in similar mitochondrial defects as the parkin gene including abnormal mitochondria, increased sensitivity to oxidative stress, and increased oxidative damage [8].
HTRA 2 Gene
It is rare to find mutations of the HTRA 2 gene in individuals with PD; however, their occurrences are thought to increase the risk for development of the disease as demonstrated via striatal degeneration and parkinsonism in homozygous knockout mice. Mutations of the gene result in increased mitochondrial volume, electrochemical changes in the mitochondrial membrane, and increased risk of cell death related to the activity of staurosporine, an ATP kinase inhibitor. It is also thought that mutations to the PINK 1 gene negatively influence HTRA 2 function [8].
Secondary Parkinsonism
Secondary parkinsonism is defined by O’Sullivan [7] as “a group of disorders with identifiable causes that produce abnormalities of basal ganglia function” and present with symptoms similar to PD [7]. Viruses, toxins, drugs, vascular disease, basal ganglia tumors, normal pressure hydrocephalus, hemiparkinsonism-hemiatrophy, and metabolic causes (hypothyroidism, hyperparathyroidism, hyperparathyroidism, Wilson’s disease).
Viruses
The influenza virus, encephalitis lethargica, created an epidemic from 1917 to 1926. Years later, a group of affected individuals developed post-encephalitic parkinsonism, a clinically and pathologically separate disease from PD. The incidence of this disease is gradually decreasing and there has been no recent recurrence of encephalitis lethargica. Thus, there are thoughts that the virus was responsible for this parkinsonian syndrome [7],[2].
Environmental Factors
Epidemiological studies have demonstrated that the following environmental factors are positively correlated with the risk of developing PD: living in a rural environment, pesticide use (ex. rotenone), contact with certain industrial chemicals (trichloroethylene, manganese, carbon disulfide, carbon monoxide, cyanide, methanol), exposure to wood preservatives, drinking well water, and exposure to MPTP [7],[2],[8],[1]. There are thoughts that some of these environmental factors contribute to the mitochondrial dysfunction seen in primary PD [8],[1]. One of the strongest relationships reported in the literature is the negative correlation between cigarette smoking and the development of PD [2],[1]. One study also demonstrated a similar inverse relationship between caffeine intake and risk of PD development [2].
Drug-induced parkinsonism (DIP)
After idiopathic PD, drug-induced parkinsonism (DIP) is the second most common cause of parkinsonism in the elderly and is often misdiagnosed as PD [10]. Some of the most common drugs involved include: typical antipsychotic agents (chlorpromazine, promazine, haloperidol, trifluoperazine, sulpiride, flupentixol, pimozide, fluphenazine, benperidol), calcium channel agents (flunarizine, cinnarizine),antiemetic agents (prochlorperazine, metoclopramide), atypical antipsychotic agents, particularly at higher doses (risperidone, olanzapine), antihypertensive agents (reserpine, a-methyldopa), and tetrabenazine [7],[10]. It is thought that these drugs interfere with presynaptic or postsynaptic dopaminergic mechanisms [7]. In the case of patients exposed to typical antipsychotic agents, also known as neuroleptics, nearly 80% develop extrapyramidal symptoms and nearly 25% develop DIP [10]. The symptoms of DIP can typically be reversed within a few weeks following termination of the responsible medication [7].
Parkinson-Plus Syndromes
Parkinsonian symptoms are one of the potential manifestations in a group of neurodegenerative diseases that affect the substantia nigra (striatonigral degeneration, Shy-Drager syndrome, progressive supranulear palsy, olivopontocerebellar atrophy, cortical-basal ganglionic degeneration, multi-infarct vascular disease, Alzheimer’s disease, diffuse Lewy body disease, normal pressure hydrocephalus, Creutzfeldt-Jakob disease, multiple system atrophy, Wilson’s disease, and juvenile Huntington’s disease) [7]. Most of these diseases are rare with progressive supranuclear palsy, multiple system atrophy, and dementia with Lewy body disease being the most common [7],[11]. Symptoms of rigidity and bradykinesia in the early disease course often make these syndromes easily confused for PD. However, additional symptoms specific to the diseases underlying etiologies eventually occur and the parkinsonian symptoms rarely improve with anti-Parkinson medications such as levodopa [7].
Clinical Manifestations
The onset of Parkinson’s disease is often subtle and very gradual. Unfortunately, because early signs/symptoms aren’t often obvious, they may go unnoticed and the disease may subsequently go undiagnosed for some time, especially if the individual does not have the characteristic resting hand tremor. This early period may last 1-2 years before more pronounced symptoms appear. There are however many hallmark signs of Parkinson’s disease. These signs and symptoms are illustrated below in Figure 3 and subsequently listed for more detail.
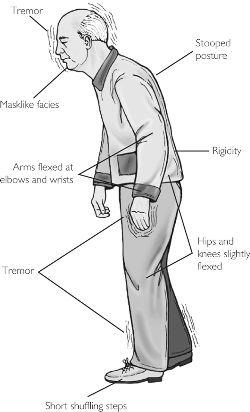
Figure 3. Classic presentation of Parkinson's disease
From: http://schoolworkhelper.net/2011/02/parkinson%E2%
80%99s-disease-symptoms inheritance/
Common symptoms [4],[20],[21]:
- Tremor
- Present when extremity/limb is at rest
- Terminates with movement
- Resting hand tremor is most characteristic (pill-rolling)
- Bradykinesia
- Rigidity of the extremities or trunk
- Impaired posture and balance
- Hypokinesia
- May present itself in facial expression, decreased arm swing with gait
- Leads to an increase in fall risk secondary to inability to generate adequate muscle force quickly and decreased ability to make postural adjustments/corrections in a timely fashion to avoid a fall
- Visuoperceptive impairments
Secondary symptoms [4],[20],[21]:

Figure 4. Micrographia seen in PD
From: http://upload.wikimedia.
org/wikipedia/commons/2/26/
Writing_by_a_Parkinson's_disease
_patient.jpg
- Micrographia
- Decreased arm swing with gait
- Foot drag resulting in shuffled gait
- Freezing
- Hypomimia
- Hypophonia
- Retropulsion
- Decrease in/loss of automatic movements (i.e. blinking, swallowing)
- Cognitive impairment (present to some extent in up to 30% of individuals)
- Memory loss
- Impaired judgment
- Poor planning
- May preceed PD dementia
- (Parkinson’s) Dementia
- Interferes with the following:
- ability to plan
- ability to maintain goal orientation
- ability to make decisions
- Interferes with the following:
Other symptoms [4],[20],[21]:
- Depression
- Pain
- Anxiety
- Psychosis (typically visual hallucinations)
- Sleep disturbances
- Autonomic dysfunction (i.e. constipation, orthostatic hypotension)
Clinical diagnosis of Parkinson’s disease requires hypokinesia affecting the upper body in combination with resting tremor and/or rigidity [4].
Disease Progression [4],[22]
While each individual with PD progresses at a different rate and may experience various symptoms in each stage, the motor progression tends to occur in predictable stages with corresponding signs/symptoms. The Hoehn and Yahr Stage scale is the most commonly used scale for discussing the severity of PD. It is described below and has since been modified with a stage 0, as well as stages 1.5 and 2.5. Parkinson’s disease is not fatal in and of itself. However, those with the diagnosis do tend to have decreased life expectancies. In the later stages of the Parkinson’s disease, increased risks for death include choking, pneumonia and severe (potentially fatal) falls.
Stage I
- Signs and symptoms are unilateral
- Mild tremor of a single limb
Stage II
- Signs and symptoms are bilateral
- Changes in posture and gait
- Minimal disability noted overall
Stage III
- Significant hypokinesia
- Generalized dysfunction (moderately severe)
- Deficits of equilibrium/balance affecting gait and standing
Stage IV
- Ambulation limited
- Rigidity
- Bradykinesia
- Unable to live alone
Stage V
- Extreme weight loss
- Spends most of the day in a wheelchair or bed
- Unable to live without assistance/requires constant supervision
- Assistance needed for ADLs, mobility, etc.
- Cognitive deficits may be present and/or prominent (i.e. hallucinations and delusions)
- Benefits of PD medications vs. PD medication side effects are considered
Finally, it is important to note that there are other types of parkinsonism that do exist and share some of the common symptoms of PD. The following list can help in distinguishing PD from parkinsonism syndromes.
- Clinical signs/symptoms consistent with other another diagnosis/disorder
- Gait and balance dysfunction present earlier rather than later in course
- Symptoms do not effectively respond to anti-Parkinson medications
- Imaging studies show involvement of other structures other than the basal ganglia
Mice and Nonhuman Primate Animal Models: MPTP
Overview

Figure 2. Mice: An Animal Model for PD.
From: http://whsc.emory.edu/home/news/img/mice_195x145.jpg
MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) was first discovered to produce a clinical syndrome similar to PD in the 1980’s when a group of young addicts using synthetic heroin were seen in emergency rooms for symptoms including tremor, rigidity, bradykinesia, and postural instability [12],[13]. As in PD, these individuals were also responsive to the drug therapy levodopa. Eventually, analysis of the heroin confirmed the presence of MPTP. Around the same time, similar syndromes were reported in a group of chemists with exposure to MPTP and also in a student injecting a homemade meperidine derivative. Neuropathological brain examination after the death of the student revealed damage to the substantia nigra. The recognition that MPTP was the common denominator in all of these cases quickly lead to the development of animal models using this toxin [12]. Mice and nonhuman primates, often the macaque, are the two most common models utilized [13],[14]. While mice are a practical model for studying neuroanatomical and neurochemical changes associated with MPTP exposure, primates are preferred for studying behavioral manifestations [13]. In addition, the primate MPTP model is considered the gold standard in drug development for PD [14]. Mice require more frequent and much higher doses of MPTP than primates as it is more rapidly metabolized in these animals [12]. In addition, a very specific strain of mouse (C57black/6) is typically used as it is most sensitive to MPTP injection [13]. In response to systemic injection, both animal models demonstrate loss of striatal dopamine and substantia nigra neurons, behavioral symptoms similar to PD, and responsiveness to levodopa therapy [12].
Mechanisms
MPTP is systemically injected into the animals and because it is a lypophyllic substance, MPTP readily crosses the blood-brain barrier [12]. In glial cells, MPTP is quickly metabolized into MPDP+ by MAO-B (monoamine oxidase B) and spontaneously oxidized into MPP+ by mechanisms that are not well understood [12]. MPP+ is then actively transported into the extracellular space and enters nearby dopamine neurons via an active dopamine transporter in the plasma membrane [12],[13],[14]. This transportation process has been coined the fatal flaw of the dopamine cells that results in their increased susceptibility to MPTP toxicity [12]. It has been found that substantia nigra neurons are more sensitive than ventral tegmental neurons to this process. Once inside the dopamine cells, MPP+ causes damage to complex 1 of the mitochondria’s electron transport chain resulting in decreased ATP formation and the production of reactive oxygen species (ROS). Consequently, the cells are unable to maintain some of their crucial energy-dependent concentration gradients. This triggers a cascade of events that lead to damage of dopamine cell lipids, proteins, and DNA and eventually result in apoptosis. It has been found that elevated concentrations of iron and melanin potentiate these toxic mechanisms. Thus, the high iron content combined with the high metabolic activity of dopamine cells is thought to contribute to their vulnerability. These processes have been found to be further exacerbated by protein mutations, age- related losses of dopamine cells, and exposure to other environmental toxins such as the pesticide rotenone [12].
Limitations
Possibly the largest limitation to the use of MPTP to create animal models of PD is the absence of Lewy bodies in response to injection, a primary pathological mechanism of the true disease. In addition, it demonstrates minimal changes to ventral tegmental dopamine neurons and brain norepinephrine and serotonin concentrations [12].
Uses
Over the past 20-plus years, the MPTP animal models have been crucial for enhancing the understanding of the neurochemical pathology and cellular mechanisms involved in PD. Furthermore, researchers have used the models to study the influence of genetics and environmental factors and increase the knowledge of the inflammatory processes involved in the disease. Future implications for these models include further exploration of risk factors, early diagnostic markers, and treatment options including neuroprotective interventions [12].
Rat Animal Models: 6-OHDA
Overview
6-hydroxydopamine (6-OHDA) is also a neurotoxin that is commonly used in rats to create an animal model of PD that has proved particularly useful for studying the effects of intervention strategies.[15] When it emerged over 30 years ago, it was the first animal model of PD to induce death of substantia nigra pars compacta (SNpc) cells. [16] As the pathology of damage in response to 6-OHDA exposure varies from PD pathology, it is not as widely used as the MPTP model.[16]
Mechanisms
To prepare the animals, the toxin must be directly injected into the midbrain as it cannot cross the blood brain barrier. Typically, the striatum, SN, or median forebrain bundle are chosen as sites of injection. [15] [16] The median forebrain bundle is utilized as it contains ascending connections from the basal ganglia to the forebrain. [16] Death of nigrostriatal neurons typically occurs within 24 hours of injection. Similar to the MPTP model, the susceptibility of these cells to damage is primarily due to the preferential uptake of the toxin by dopamine and noradrenaline cell transporters. Once inside the cells, 6-OHDA collects in the cytosol and induces oxidative stress and production of quinones. These compounds participate in events leading to cell death including the inactivation of proteins and nucleic acids.[16] The degeneration of affected neurons occurs in a retrograde fashion and leads to morphologic changes similar to apoptosis. There is also evidence of surrounding gliosis after 6-OHDA exposure.[15] The toxin is often injected unilaterally to allow for the use of the contralateral brain as a control. This preparation causes animals to adopt an asymmetrical circling behavior that worsens as severity of the lesion increases. This model, therefore, is particularly useful for studying treatment effects. [16]
Limitations and Uses
Similar to the MPTP models, one of the major limitations of this model is the lack of Lewy body inclusion. When compared to animals exposed to MPTP, those exposed to 6-OHDA demonstrate dopamine loss that is more severe and longer in duration.[15] [17] It has also been stated that MPTP models may better represent earlier stages of PD versus 6-OHDA models.[17]
Inflammation and Immunity in PD
Microglia, a component of innate immunity, monitor the environment of the central nervous system [48]. When activated, they multiply, divide and subsequently increase their cell volume [48]. Activated microglia are a large component of inflammation and subsequently lead to an increase in complement receptors and cell adhesion molecules such as lymphocyte function associated antigen (LFA-1), intercellular adhesion molecule (ICAM)-1 cluster of differentiation (CD) 54, vascular cell adhesion molecule (VCAM)-1 (CD 106) and CD1 [50]. Furthermore, increased expression of ICAM-1 and its counter-receptor, LFA-1, has been shown [51]. Both have shown masses of reactive microglia as well as dispersion of LFA-1 leukocytes present throughout PD brains both in humans and animal models [51].
Activated microglia secrete various substances that induce death to agents that are harmful to the body but, in doing so, have consequences for neuronal injury/death, an underlying factor in the pathophysiology of PD and thus implicating their role in the disease [48]. These substances include reactive oxygen species, reactive nitrogen species, proinflammatory cytokines and prostaglandins. [48]. Thus, in the instance that the activation of microglia is not tightly controlled it will subsequently result in toxicity and possibly death to neurons secondary to the release of pro-inflammatory factors including interleukin one beta, tumor necrosis factor alpha, IL-6, nitric oxide, prostaglandin E2 and superoxide radical [48]. Furthermore, the substantia nigra is particularly vulnerable to dysfunctional microglial activation because microglia is populous in the brain [48]. Unusually high levels of interleukin one beta and tumor necrosis factor alpha have been noted in peripheral and CNS tissues in PD[52],[53]. Furthermore, both in vivo and in vitro studies have shown that microglia are activated by the overexpression of alpha synuclein, a constituent of the Lewy body inclusions commonly found in the brains of those with PD. [57], [58].
T cells and B cells, part of the adaptive immune system, flood to sites of inflammation and trigger the immune response when activated. Typically, they are not able to pass into the central nervous system (CNS). However, in a neuroinflammatory state, activated glial cells release substances that disrupt the blood-brain barrier and thus allow such cells to pass into the CNS. In the brains and CNS tissue of those with PD, T-cells have been shown to be widespread throughout, supporting the role of inflammation in the disease [51]. Furthermore, in a study that transferred splenocytes into mice with PD (MPTP-induced) demonstrated much infiltration throughout the brain as well as the substantia nigra (for reasons as mentioned previously) [((bibcite Benner)]. There is evidence that nitrated alpha-synuclein activates leukocytes in the periphery [((bibcite Benner)]. The death of dopaminergic neurons (a major component in the pathophysiology of PD) has been shown to be increased and worsened by T cells toward nitrated alpha-synuclein [59]. With all this evidence and information taken together, again a link between the inflammation and activated microglia and PD pathophysiology can be surmised [59].
There are a few studies implicating involvement of the immune system in PD as well as evidence to support its role. The immune system’s role in regulating homeostasis may underlie the mechanism behind the activation of microglia and the subsequent effect on dopaminergic neurons [48]. Furthermore, there are various sources of oxidative stress involved with Parkinsons disease [48]. Oxidative stress may be the possible “trigger” for the inflammatory response associated with the death of dopaminergic neurons [48]. Additionally, while studies shown infiltration of adaptive immune cells in the brain, there have been others that have shown presence of other inflammatory cytokines [((bibcite Stone). In the substantia nigra, the cite of dopaminergic neuronal death associated with PD, studies have shown that higher numbers of T cells are found very near to activated microglia and degenerating neurons [46]. Furthermore, in a study with PD patients, the total numbers of lymphocytes were decreased by 17%, while specifically CD 19+ B cells and CD 3+ T cells were decreased by 35% and 22% respectively [60].
One study supports this with a finding of a possible genetic association with PD in the human leukocyte antigen (HLA) region [45]. This evidence is suggestive of the significant involvement of neuroinflammation and humoral immunity [45]. Other studies have shown presence of reactive DR-positive microglia in the substantia nigra of individuals with PD [46] as well as animals affected with MPTP (see Animal Model Section) [47].
Natural regulatory T cells are also of importance to mention when discussing inflammatory processes and PD. They function to “prevent immune responses toward self-peptides”. They aid in controlling reactivity of the innate immune system and down regulating the responses of CD4+ and CD8+ T cells and B cells through production of anti-inflammatory cytokine (i.e. IL-10, TGF-beta) [63],[64],[65]. Furthermore they work to down regulate the immune responses by cytolysis [66] and metabolic disruption [67]. Regulatory T cells inactivate effector cells (B cells, T cells, myeloid, microglia) [70],[71],[72].
Pathophysiology

Figure 5. Pathophysiology of Parkinson's Disease
From: http://www.cellsignal.com/reference/
pathway/images/Dopamine_Parkinsons.jpg
Neuronal changes in Parkinson’s disease are widespread, however the principal neurological abnormality that occurs is degeneration of dopamine-producing cells in the Substantia nigra pars compacta (SNpc) portion of the basal ganglia. In addition to this neuronal loss, formation of ubiquitin-positive cytoplasmic inclusions called Lewy bodies occurs in remaining neurons [18]. Nigrostriatal neuronal cell bodies are located in the SNpc, and project mainly to the putamen. In a normal state, these neurons contain evident amounts of neuronmelanin, neuropathological finding of SNpc depigmentation. This SNpc depigmentation has been shown to correspond with the amount of expression of the DA transporter mRNA, and supports the finding that DA depletion is greatest in the dorsolateral portion of the putamen, the primary project site for these neurons [16]. In a normal, healthy individual, the neurotransmitter dopamine is released in the presynaptic neuron, and through activation of D1 and D2 type dopamine receptors in the postsynaptic neuron, G proteins signal activation of adenylate cyclase. This in turn results in cAMP formation (a second messenger) and activation of PKA (Protein kinase A). Dopamine-producing cell death combined with a breakdown in the cellular pathway above results in a decrease of dopaminergic input to the corpus striatum. Decreased dopaminergic input to the corpus striatum appears to be associated with motor manifestations of PD. At the onset of motor symptoms, putamen dopamine is depleted by approximately 80%, and about 60% of SNpc neurons have already been lost [16]. This clinical-pathologic correlation is supported by findings that the N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is selective for dopaminergic neurons in the substantia nigra in humans and primates, but produces the full range of clinical motor symptoms seen in PD [19].
The significant neuronal change which occurs in PD is not exclusive to dopaminergic neurons. Neuronal cell death and LB formation is also found in noradrenergic (locus coeruleus), serotonergic (raphe), and cholinergic neurons, as well as the cerebral cortex (in particular cingulate and entorhinal cortices), olfactory bulb, and the autonomic nervous system [16]. In addition to motor symptoms, dementia occurs at a high rate in those with PD, and can be attributed to degeneration of hippocampal structures and cholinergic inputs [16]. These extensive neuronal changes have far-reaching effects on neurotransmitter release in the brain. Current research suggests that other neurotransmitter imbalances may also be present following dopamine loss in the basal ganglia, including gamma-aminobutyric acid (GABA), 5-hydroxytryptoamine (serotonin), endogenous opioids, and excitatory amino acids (glutamate). Outlined further in the Etiology section here, predisposition by familial genetic mutation or exposure to environmental or neurotoxins can result in a cascade of cellular responses that include apoptosis and the misfolding of alpha-synuclein, which has been attributed in the formation of Lewy bodies.
According to Dauer and Przedborski, current literature supports two main theories for pathogenesis of PD [16]. One hypothesizes that misfolding and aggregation of proteins are influential in the loss of SNpc dopaminergic neurons, while the other is based on the idea that irregular mitochondrial structure and function contributes to increased oxidative stress and toxic dopaminergic species. These two pathological mechanisms are not mutually exclusive, however, and current PD research aims to paint a clearer picture of how these pathways may contribute to the provocation and progression of PD, and how they may possibly interact. Currently known about the first hypothesis regarding abnormal deposition of protein in brain tissue is that oxidative damage to alpha-synuclein contributes to its ability to misfold and aggregate. This could potentially be neurotoxic through a variety of mechanisms. Protein aggregates could cause direct structural damage or interfere with intracellular processes. Protein inclusion may also cling to proteins for cell survival [16].
Although age cannot be denied as a contributing factor for PD, loss of dopaminergic neurons seen in PD can be distinguished from normal changes associated with aging. In those with PD, neuronal cell death occurs mainly in ventrolateral and caudal portions of the SNpc; in normal aging these changes are seen in the dorsomedial aspect of the SNpc. PD is diagnosed clinically in life, but definitive diagnosis cannot be made with detection of SNpc dopaminergic neuron loss and the presence of Lewy bodies [16].
Lewy Bodies
Structure
One of the pathological hallmarks of PD, Lewy bodies (LB) are spherically-shaped eosinophillic cytoplasmic protein aggregates found in all affected brain regions that are made up of several proteins, including α-synuclein, parkin, ubiquitin, and neurofilaments. [16] Greater than 15 µm in diameter, LB’s have an organized structure which contains a dense hyaline core framed by a clear halo. [16] When examined in greater detail through the use of electron microscopy, a dense granulovesicular core bordered by a ring of 8-10 nm fibril extensions is revealed. [16] Immunohistochemical staining with an antibody against α-synuclein shows a LB with an extremely immunoreactive central zone surrounded by a slightly immunoreactive peripheral zone. Immunostaining with an antibody against ubiquitin displays greater dispersion of immunoreactivity throughout the LB. [16]
Presence and Role in PD
Current diagnosis of PD in life is made through identification of clinical signs and symptoms, but definitive neuropathological diagnosis can only be made with detection and quantification of Lewy bodies. [16] While LB’s are found in all affected brain regions of those with PD, they are mainly found at the major sites of neuronal death, i.e. the substantia nigra and locus coeruleus. [23] However, it has been shown that LB presence in the substantia nigra does not predispose those particular neurons to apoptosis more so than general substantia nigra neurons, and that a majority of the neurons that do undergo apoptosis do not contain LB’s [24] Attempts to correlate either cortical or brain stem LB density with disease duration, early onset, presentation at onset, presence or absence of cognitive changes, visual hallucinations, delusions, persistent falls, or severity of symptoms have been unsuccessful. [25] [26] [27] [28] One study did demonstrate weak evidence for a correlation between the density of LB’s in the cingulated gyrus and cognitive decline. [27]
In PD, LB’s contain α-synuclein that has been altered by oxidative activity. In-vitro studies have shown to this form of the protein demonstrates a greater tendency to aggregate than unmodified α-synuclein, [16] which supports the theory that oxidative stress plays a key role in PD pathogenesis.
LB formation is found not only in PD, but in other neurodegenerative diseases such as Alzheimers Disease (AD) and incidentally as a pathological finding in advanced aging. The explanation for this increased frequency of LB’s seen in AD and their overall role in neuronal cell death is a topic of debate. Examination of the brains of asymptomatic individuals show that the occurrence of LB’s increases with age, which raises the question of whether LB formation predisposes PD symptoms or is a feature of normal aging. [23] In a review of 904 autopsies, 106 individuals were found to have LB formation in the brain, but only 32 of those had been diagnosed with a neurodegenerative disease. [29] A study by Gibb and Lees also found a positive relationship between age and occurrence of LB’s from 3.8% to 12.8% between the sixth and ninth decade of life, which surpasses the incidence of PD cases by 3 to 6 times. In summary, there is currently insufficient evidence to justify the role of LB’s in the pathological process of PD, although their presence continues to be a pathological hallmark of diagnosis. [23]
Cellular Components with Related Function in Pathogenesis of PD
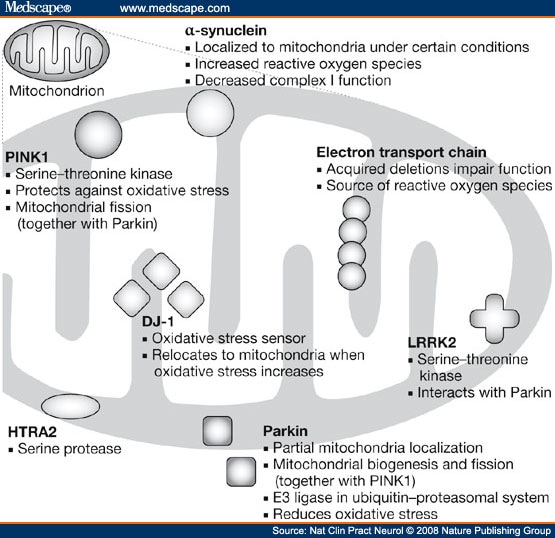
Figure 6. Important Cell Components in PD and Corresponding Function.
From: http://img.medscape.com/fullsize/
migrated/583/365/ncpn583365.fig2.jpg
Mitochondria
- The mitochondria are complex organelles with several functions critical to optimal cellular functioning. The crucial functions of the mitochondria under normal conditions include the following: synthesis of ATP through oxidative phosphorylation, oxidation of fatty acids, regulation of redox state and cell calcium signaling, as well as control of apoptosis in response to both intracellular and extracellular events [30]. Mitochondria have an inner and outer membrane that functions to separate the inner mitochondrial matrix from the intermembrane space, and the intermembrane space from the cytosol of the cell as a whole [30]. The inner membrane is highly folded into cristae; the cristae contain the respiratory chain complexes of the electron transport chain along with ATP synthase [30]. The mitochondria are able to alter their shape, size, and inner membrane structure in response to metabolic stimuli as well as external signaling pathways [30]]
- Within individual neurons, the mitochondria are very abundant at synapses, and in unmyelinated axons of the peripheral nervous system [30]. Mitochondria concentrate and are recruited to subcellular regions with high metabolic requirements, such as active growth cones in neurons. Mitochondria regulate neuronal activity, synaptic plasticity and neurotransmitter release by controlling synaptic calcium concentrations as well as ATP-dependent vesicle movement [30].
- Studies have recognized the role of Parkin and PINK 1 in abnormal mitochondrial function and dynamics, leading to DA neurodegeneration within the striatum [30]. Parkin functions to maintain overall mitochondrial integrity and protects the cell against both oxidative stress and stress from unfolded proteins; PINK1 acts as a protein kinase also to protect the cell against oxidative stress [30]. In mice, a compete deletion of the PINK1 gene resulted in decreased DA release and notable mitochondrial defects within neurons of the striatum. Additionally, PINK1 mutation results in dysregulation of mitochondrial calcium homeostasis, which further results in calcium overload inside the cell and increased production of reactive oxygen species (ROS) [30]. PINK1 kinase activity to activate Parkin increases upon activation of certain stress-signaling pathways [30]. As a result, Parkin is recruited to mitochondria under conditions that might harm these organelles [30]. In short, with mutations in either of these genes, the autophagy system would be overwhelmed by the mass of defective mitochondria, and neuron death would result. This is the suggested pathogenesis of PD.
- Mitochondria and neuronal transmission. Dysfunctional calcium homeostasis in PINK1 deficient neurons and impaired calcium storage has been shown to block mitochondrial transport and result in decreased energy supply at neuron synapses [30]. This has been the explanation for abnormal DA transmission in mice with PINK1 deletion because mitochondrial dynamics and axon trafficking of mitochondria are important for synaptic functional and neurotransmission [30].
- Mitochondria and DA neurons. Mitochondrial density and mass are shown to be lower in the DA neurons of the SNpc when compared to non-DA neurons [30]. Additionally, cytoplasmic DA is prone to auto-oxidation and natural oxidation from enzymes in the cytosol [30]. This oxidation can lead to impaired mitochondrial respiration and membrane permeability [30]. DA has also been shown to interfere with autophagy, and to promote cell death via upregulation of alpha-synuclein expression [30]. Therefore, if DA is not sequestered properly into cellular transport vesicles, DA has the capacity to affect mitochondrial respiration and transport. In addition, through its effects on autophagy, DA may interfere with clearance of aggregated proteins and dysfunctional mitochondria [30]. Interestingly, DA has been shown to modify Parkin in living DA cells through covalent bonding; and this has led to decreased Parkin ligase activity [30]. This suggests that DA-dependent de-activation of Parkin has the capacity to contribute to loss of DA-containing neurons, which may lead to further mitochondrial dysfunction and impaired mitophagy. So overall, this demonstrates that DA and its metabolites can negatively affect mitochondrial homeostasis and optimal functioning, further rendering DA neurons more susceptible to impairments of mitochondrial function and clearance.[30]
PINK1
- PINK 1 gene encodes a putative serine/threonine protein kinase with a mitochondrial import sequence located at its N-terminus [35]. PINK 1 is expressed in all adult human tissues, and is also present in certain areas of the brain [35]. This protein is processed inside the mitochondria to generate two mature proteins. PINK 1 interacts with Parkin to regulate mitochondrial dynamics, including fission/fusion. PINK 1 recruits Parkin to damaged mitochondria in response to stress-signaling pathways, thus assisting to regulate the degradation of mitochondria through autophagy and attenuate oxidative stress. [35]
- PINK 1 initiates mitophagy via phosphorylation of Parkin [36]. PINK1 stabilization on the outer mitochondrial membrane is a necessary for Parkin ligase, which is normally localized to the cytosol, to translocate to damaged mitochondria [36]. PINK1 mutations that are related to PD pathology impair mitochondrial autophagy by compromising initial Parkin translocation to mitochondria; in normal circumstances, PINK 1 recruits Parkin following a loss of mitochondrial membrane potential [36].
HtrA2
- This is a second substrate (along with Parkin) acted on by PINK1, and is a mitochondrial protease [30]. PINK 1 phosphorylates HtrA2 in response to the activation of the p38 stress-signaling pathway which then results in increased HtrA2 protease activity. Increased HtrA2 protease activity may regulate neuron survival under stress conditions [30]. Neurons in the striatum of HtrA2 knockout mice are degenerated, and subsequently results in a disorder with PD phenotype [30].
DJ-1
- The DJ-1 gene encodes a protein that is normally involved in every-day cellular processes such as sperm maturation, fertilization, and oncogenesis [37]. It may also have the following actions within the cell: peroxidase, chaperone, and a regulator of transcription or RNA metabolism [37]. DJ-1 is normally localized in the cytosol, but it is localized to the mitochondria under conditions of oxidative stress and has been found to play a significant part in the defensive cellular response to oxidative stress [37]. It is also suggested that this could work upstream of the PINK1/parkin activation or it could work within an independent, but parallel, pathway in conjunction with PINK1/Parkin.
- DJ-1 knockout mice did not have neuron death; however, they did demonstrate increased vulnerabilities to neurotoxic agents and increased sensitivity to oxidative stress [37]. DJ-1 deficient cells also have lower mitochondrial fusion, an increased tendency to fragment, and impaired mitochondrial dynamics leading to an increase in autophagy. This also suggests that DJ-1 function in normal cells could regulate autophagy or attenuate the downstream negative effects of reactive oxygen species [37].
LRRK 2 (leucine-rich repeat kinase 2 gene)
- LRRK2, also called leucine-rich repeat serine/threonine-protein kinase 2, is localized in the cytoplasm. Although its actual role is unknown, it may take part in phosphorylation of proteins or in GTPase activity. Additionally, it interacts with PARK2 (Parkin). Mutations of LRRK2 cause PARK8 (familial Parkinson's disease, type 8) and augment MAPK activity. It causes an increase in phosyphorlation intra and inter-cellularly. [38]
- LRRK2 is associated with idiopathic PD. It is typically late-onset PD, but the overall clinical phenotype is not different from other types of PD.[39] It is inherited through autosomal dominant inheritance. Mutations have been found to be more common with a family history of LRRK2 PD.[39] It is a gene with 51 exons, but screening efforts have focused on exons 19, 24, 31, 35, 38, and 41.[39] Sporadic and familial mutations have both been found. Some mutations may not be fully penetrant (important for diagnostic screening).[39] The G2019S mutation is the most common. [39]
- The protein product, containing 2527 amino acids, is dardarin. The gene is located within the PARK8 locus on chromosome 12p11.2-q13.1. [40]
- Individuals may have Lewy bodies, Lewy neurites, or protein deposits (mostly in the cytoplasm). The pathology of LRRK2 includes deposits of aggregated protein.[41] A study using CD-1 outbred mice demonstrated that aggregates containing LRRK2 and ubiquitin are increased when LRRK2 is expressed with parkin.[41] LRRK2 in its mutated form affects SH-Sy5Y cells and primary neurons causing neuronal degeneration.[41] LRRK2 has been found to interact with parkin (in the RING2 domain of parkin; the same location for interaction with synphilin-1) but not alpha-synuclein, DJ-1, or tau.[41] This in-vitro study did not find that LRRK2 would be directly ubiquitinated by parkin. LRRK2 was found to cause a 25-fold increase in autoubiquitination by parkin.[41] Mutant LRRK2 caused more cell toxicity than the control in SH-SY5Y cells and mouse primary neurons.[41] Neuronal cell viability was decreased and there was apoptotic cell death in the SH-SY5Y cell. Parkin was not found to protect against cell toxicity. [41]
PARK 2 gene (Parkin)
- The protein encoded by the Parkin gene is an E3 ubiquitin ligase that catalyzes polyubiquitination of mis-folded or defective proteins and directs them to the proteosome to be degraded [32]. Parkin, which is normally in the cytosol (as mentioned above), translocates from to the mitochondria in response to a fall in mitochondrial membrane potential [30] [32]. A fall in membrane potential results in phosphorylation of parkin by PINK 1 to activate the protein ligase [32]. Cellular Parkin can also be phosphorylated through Cdk5, in addition to PINK 1 [32].
- A loss-of-function mutation in this gene results in early-onset PD, which is recessive [32]. Parkin knockout mice have striatal mitochondria with lowered respiratory capacity, as well as notably increased oxidative stress [33]. Additionally, both mitochondrial function and morphology are impaired in fibroblasts from patients with Parkin mutations, and the mitochondrial dysfunction subsequently results in accumulation of defective mitochondria [33] [32]. Interestingly, Parkin also determines the intrinsic cellular threshold for the release of cytochrome c from the mitochondrial matrix. Additionally, the levels of cellular Parkin expressed inversely correlated with cytochrome c release and apoptosis, meaning that increased expression of Parkin to regulate oxidative stress results in decreased cytochrome c release and decreased apoptosis [33]. Parkin also attenuates activation of p53, which is a tumor suppressor gene that has the capacity to activate apoptosis [33]. The transcriptional expression of mono-amine oxidase (MOA), which can generate reactive oxygen species and eventually kill DA neurons, can be repressed by Parkin activity; which suggests a protective mechanism that is lost in patients with PD who have a mutated Parkin protein. [33]
- Although the parkin knockout mice themselves do not demonstrate all phenotypic features of PD-related PARK2 in humans, these mice do have increased vulnerability toward inflammatory neurotoxic stimuli, an accumulation of mitochondrial abnormalities, and increased oxidative stress [33].
Alpha-synuclein
- This protein is a major component of the Lewy body and localizes normally to presynaptic terminals of neurons [32]. While the exact physiological function is poorly understood, current research has suggested it has a role in dopamine synthesis, synaptic plasticity, and vesicle dynamics [32]. Abnormally high levels of this protein due to unbalanced protein production and/or degradation are thought to cause death of DA neurons in patients with PD [32]. Viral vector-mediated overexpression of this protein in rodents has been shown to cause degeneration of DA neurons and subsequent apoptosis [32].
- Several post-translational modifications have been reported to alpha-syn in patients with PD, such as phosphorylation or truncation [32]. In the drosophila model of overexpression, a phosphorylation at site Serine-129 resulted in higher DA neurotoxicity and exacerbated DA loss within the neuron [32]. However, there is conflicting evidence as is has been reported that the alpha-syn induced death of DA neurons was unchanged by the phosphorylation site or status [32]. On the other hand, the viral vector-mediated alpha-syn delivery does cause an abnormal accumulation of the alpha-syn protein, and results in subsequent degeneration of SNpc DA neurons in both mature rodents and primates [32]. Evidence also demonstrates that DA cell death from alpha-syn occurred through activation of caspase-9 and caspase-3 [32]. It is also suggested that both PINK1 and Parkin can suppress the toxicity of alpha-syn and prevent accumulation in animal models of PD [32].

Figure 7. Role of Cell Components in Neurodegeneration.
From: http://www.hih-tuebingen.de/typo3temp/pics/efe62d44bf.jpg
Ubiquitin Ligase
- This is a protein that leads to the attachment of ubiquitin molecule to the lysine of a target protein. The ligase targets specific protein substrates for degradation by proteosomes in response to stress-signaling pathways that have been activated [31].
Proteosome
- Proteosomes are large protein complexes located within both the nucleus and the cytoplasm of all cells [31]. The primary function of the proteosome is to degrade damaged proteins by a chemical process known as proteolysis. Proteolysis is a chemical reaction characterized by the breaking of peptide bonds, and is carried out by enzymes within the protein complex called proteases. This process yields peptides that can be further broken down into amino acids and then used to synthesize new proteins [31].
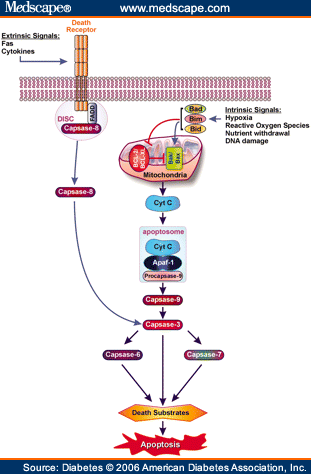
Figure 8. Role of Cytochrome c, Caspase -3 and -9 and Reactive Oxygen Species in Apoptosis.
From: http://img.medscape.com/fullsize/migrated/540/528/db540528.fig1.gif.
Cytochrome c
- This is a small heme (iron-containing) protein associated with the inner membrane of the mitochondria [31]. This protein is a component of the electron transport chain; it accepts electrons from the b-c1 complex and transfers those electrons to the cytochrome oxidase complex. Additionally, it also is involved in the initiation of apoptosis when released into the cytosol. Once released into the cytoplasm, it binds to an apoptotic activating factor [31].
Reactive Oxygen Species (free radicals)
- These are chemically reactive molecules that contain oxygen, and are highly reactive due to the presence of unpaired electrons in their outer valence shell [31]. ROS form as a natural byproduct of the normal metabolism of oxygen, but levels can increase in times of stress. These free radicals are commonly produced in the mitochondria [31]. As a result of the electron transport chain, oxygen is normally reduced to water; however, on occasion oxygen is instead prematurely reduced and results in the superoxide radical. Mitochondrial dysfunction can lead to an imbalance between free radical production and removal, resulting in their accumulation and increased oxidative stress to the cell [31]. DA neurons exhibit hallmarks of oxidative stress in PD, and formation of Nitric Oxide (NO) has been implicated in the death of DA cells [31]. The formation of NO and reactions with other anions leads to protein oxidation and DNA damage. NO production from glial cells surrounding and normally supporting neurons can also contribute to this PD pathogenesis (ROS) [31].
Caspase-3 and Caspase-9
- Caspase proteases are a family of cysteine proteases that are crucial in the process of apoptosis. There are 2 types of caspases: Initiator and effector caspases [34]. Caspase-9 is an initiator caspase, which cleaves the inactive pro-forms of effector caspases to activate them [34]. Caspase-3 is an effector caspases, which cleaves other protein substrates in the cell to trigger the apoptotic process and has been demonstrated to be instrumental in loss of dopamine cells in PD [34].
TRAP1
- This is a third substrate for PINK1, and its phosphorylation by PINK1 is necessary to protect cells against oxidative stress-induced cytochrome c release and apoptosis [30].
Kinase
- This is a type of enzyme that transfers phosphate groups from high-energy donor molecules, such as ATP, to specific substrates. Protein kinases are used to transmit cellular signals and control inter-cellular and intra-cellular processes [31].
Glial cell line-derived neurotrophic factor (GDNF)
- GDNF is a neurotrophic factor, having neuroprotective effects on catecholaminergic central neurons, dopamine, and noradrenergic neurons [42]. It is a protein produced by striatal neurons and functionally maintains adult nigostriatal dopamine neurons. GDNF is a member of the transforming growth factor-b (TGF-b) superfamily (including three homologues: neurturin, persephin, and artemin).[42] Areas in the human brain with high GDNF expression levels are the striatum, accumbens nucleus, medial septal nucleus, anteroventral thalamic nucleus (ventrolateral part) and the anteromedial thalamic nucleus.[42] GDNF expression is stimulated by dopamine, adenosine and inflammatory signals [42].
- GDNF signals through Grfa1/RET pathways and N-CAM receptors (in the absence of RET) [42]. It can activate molecules to promote antioxidant defense and neuronal survival through varying mechanisms including inactivating apoptotic proteins. Treatment with GDNF can has been found to prevent neurotoxic damage to midbrain dopamine neurons in C57/B1 mice [42]. Ablation of the gene in GdnfK/K mice caused renal agenesis and the absence of the enteric plexus.[42] GDNF-null mice have been studied to learn the effect of the GDNF expression during adulthood. These studies illustrate the requirement of GDNF for dopamine and noradrenergic neurons to survive in the adult brain.[42]
- Current studies involve testing the therapeutic effects of GNDF in advanced Parkinson's Disease patients. See Interventions Section. [42]
- Infusion and viral-mediated delivery of GDNF, as well as transplantation of GDNF-producing cells, afford substantial neuroprotection in rodents and primate models of PD induced by MPTP. However, in rat models, GDNF overexpression does not protect nigral neurons against α-synuclein-induced toxicity, as the process of aggregation is unaffected by GDNF [43].
Brain-Derived neurotrophic factor (BDNF)
- Brain-derived neurotrophic factor (BDNF) is a natural made protein that is responsible for survival and maintenance of neurons in the nervous system. [78] In Parkinson’s disease BDNF has been shown to aid in the survival of dopaminerigic neurons in the substantia nigra. [78] BDNF is made in the endoplasmic reticulum and can be found in the brain as well as the periphery. Specifically, It can be found in many cortical, subcortical and spinal cord areas, including the substantia nigra. [78] BDNF is originally made in a pro form. When a neuron fires pro-BDNF is released and then it is spit by plasmin or metalloproteinases (MMP) in order to become mature BDNF. [78] In general MMP’s are enzymes that have a role in splitting proteins. [79] There are several forms of MMP and they can be found throughout the body including the SN. [79] In the brain MMP’s can function to increase synaptic activity. Studies have shown that in PD MMP 2 is decreased and its inhibitor TIMP-1 is increased. [79] The two receptors that BDNF binds to are tropomyosin-related kinase type B (TRKB) and the p75 receptor. [78] TRKB is found in many area of the brain such as the cortex, hippocampus, and brainstem regions including the SN and functions to promote the survival of neurons. [78] The p75 receptor is found mainly in the basal forebrain and when activated it promotes apoptosis. [78] Interestingly, pro BDNF tends to bind more with the p75 receptor and mature BDNF tends to bind more with TRKB. [78] In patients with Parkinson’s disease studies have found that BDNF and its mRNA is decreased, specifically in the neurons of the substantia nigra. [78] Furthermore, A study by howells et all found that BDNF is expressed less in the substantia nigra in patients with parkinsons. [80] This study concluded that this is due to the fact that there are less BDNF producing dopaminergic cells and the existing dopaminergic cells are also not effectively producing BDNF.
Along with affecting motor function through its presence in the neurons of the basal ganglia, BDNF also influences learning and memory through its role in long-term potentiation and synaptic growth. [78] Long-term potentiation is an increase in synaptic transmission between nuerons and is a mechanism for how learning occurs. Studies have found that a BDNF polymorphism Val66met correlates with cognitive issues in patients with parkinson’s disease. [81] In humans the BDNF gene codes for the BDNF protein. The Val66met polymorphism occurs when valine and methionine are switched in the gene sequence. [81] This mutation occurs in apoximately 30% of of the population. [81] In stroke patients this polymorphism has been shown to decrease motor learning and the release of BDNF. [83] It has been found to affect BDNF at the pro level, resulting in less pro-BDNF to be released when the neuron fires. [82] This in turn causes a decrease in mature BDNF. This mutation of BDNF has been related to cognitive problems including memory loss and reduced function of the hippocampus. [82] Studies have also shown that patients with parkinsons who have increased Val66met score lower on the MMSE. [82]
Coenzyme Q 10 (CoQ10)
- Coenzyme Q 10, also known as ubiquinone, is an endogenously synthesized lipid. Its primary function is to shuttle electrons from Complexes I and II of the mitochondrial electron transport chain to Complex III. Additionally, coenzyme Q 10 can exist in a reduced state, which is known to have antioxidant properties and has been found to protect membrane lipids as well as mitochondrial DNA from oxidative damage [44]. Studies report a decrease in coenzyme Q 10 in the brain of patients with PD [44]. As an anti-oxidant treatment, CoQ10 was found to improve cerebral metabolism as measured on MRI [44].

Figure 9. Genes Implicated in Parkinson's Disease and Relation to Mitochondria [73]
Interventions Overview
Medical and pharmacological treatments for Parkinson’s disease are limited to the symptomatic relief of patients, and has failed to prevent or slow down the process of neurodegeneration [77]. Due to the fact that dopamine deficiency is the main cause of PD, pharmacological treatments have been aimed at restoring the neurotransmitter levels of dopamine. Current research is aimed at developing treatments that have the potential to slow down or stop the neurodegenerative process, rather than simply treating the symptoms [75]. Other treatments for PD involve surgical techniques and deep brain stimulation.
Levodopa
The underlying pathology in PD is a deficiency in dopamine in the basal ganglia so it was originally hypothesized that a substitution of dopamine would be an effective treatment [77]. However, dopamine does not cross the blood-brain barrier and as a result is ineffective as the majority of the drug is converted in the periphery. The immediate precursor to dopamine, dihydroxphenylalanine (levodopa) is able to cross the blood brain barrier through active transport. Once levodopa enters the brain it is transformed into dopamine by decarboxylation from the enzyme dopa decarboxylase (Figure 6) [77]. Levodopa is usually taken orally and is given a peripheral decarboxylase inhibitor such as carbidopa [76]. The inhibitor prevents levodopa from prematurely being converted to dopamine in the periphery before it reaches the brain (Figure 6) [77]. Any amount of levodopa that is converted to dopamine in the periphery will not be able to cross the blood brain barrier. Although levodopa therapy is considered the gold standard for PD treatment there are many side effects and difficulties with levodopa treatment.
Levodopa Side Effects
Levodopa has been associated with gastrointestinal problems, cardiovascular issues, dyskinesias, and diminished responses [76],[77]. One of the most serious problems in levodopa treatment is that the drug seems to lose its effectiveness if given for prolonged periods (continuously for 3-4 years) [77].
Dopamine Agonists
Dopamine agonists are also used in the treatment of PD. These drugs have a similar function to dopamine and are often used in conjunction with levadopa [77]. Bromocriptine (Parlodel), pergolide (Permax), and ropinirole (Requip) are a few dopamine agonists that are used to treat PD and do not have excessive adverse effects [77].
Dopamine Agonist Side Effects
Dopamine agonists may produce nausea and vomiting and in some patients postural hypotension is also a problem [77]. With prolonged use, these drugs can cause CNS-related side effects such as confusion and hallucinations [77].
Anticholinergics
Another form of pharmacological treatment for PD comes in the form of anticholinergic drugs. The deficiency of striatal dopamine causes excessive activity in certain cholinergic pathways in the basal ganglia so drugs that limit acetylcholine transmission have been found to alleviate PD symptoms including tremors and rigidity [77].
Anticholinergic Side Effects
Anticholinergics are associated with side effects including mood change, confusion, hallucinations, drowsiness, and cardiac irregularity [77]. Other side effects are blurred vision, dry mouth, constipation, and urinary retention [77].
Other Pharmacological Agents
Amantadine
Amantadine is thought to work by blocking the N-methyl-D-aspartate (NMDA) receptor in the brain, thereby inhibiting the effects of excitatory amino acids such as glutamate [77]. This is evidence that excitatory neurotransmitters play a role in motor complications associated with advanced PD.
Amantadine Side Effects
The primary adverse side effects of Amantadine are orthostatic hypotension, CNS disturbance, and patches of skin discoloration on the lower extremities [77].
Selegiline
Selegiline (Deprenyl,Eldepryl) is a drug that strongly and selectively inhibits the monoamine oxidase type B (MAOB) enzyme [77]. This enzyme is responsible for breaking down dopamine [77].
Selegiline Side Effects
Some side effects are dizziness, sedation, gastrointestinal distress, and headache [77].
Catechol-O-Methyltransferase inhibitors (COMT)
COMT inhibitors work to prevent levodopa conversion in peripheral tissues, allowing more levodopa to reach the brain [77].
COMT Side Effects
The primary problem associated with COMT inhibitors is an initial increase in dyskinesias [77]. Other side effects include nausea, dearrhea, dizziness, and muscle pain/cramps [77].
Neurotrophic Factors
Although existing treatments are not able to stop or slow down neurodegeneration of the nigrostriatal dopaminergic pathway, current research is focused on neuroprotective agents such as neurotrophic factors (NTF) [75],[74]. NTF are secreted proteins that play a critical role in the maturation and survival of neurons. Specific NTF act in the adult brain to support and protect mature neuronal populations [75]. One NTF that has selective effects on dopaminergic neurons is glial cell line-derived neurotrophic factor (GDNF). GDNF has been shown to induce the dopamine synthetic enzyme, tyrosine hydoxylase, in fetal human rat cortical cultures [75]. It has also been shown to promote the survival and differentiaton of dopaminergic neurons in vitro and protect these cells from the dopaminergic toxins, 1-methyl-4phenylpyridinium ion (MPP+) and 6-hydroxydopamine (6-OHDA) [75]. In normal adult rats, a single injection of GDNF into either the substantia nigra or striatum significantly increased the levels of dopamine in the striatum or nigra [75]. Because GDNF does not cross the blood brain barrier, direct intracerebral delivery must be used in clinical trials. Clinical trials using direct administration to brain parenchyma using a catheter system showed good results. One study reported improvement in motor symptoms and ADL’s without serious side effects [75]. Other NTF being studied for the treatment of PD include mesencephalic astrocyte-derived neurotrophic factor (MANF), cerebral dopaminergic neurotrophic factor (CDNF), neurturin, and Coenzyme Q-10 [75],[74]. A pilot trial evaluated Co-Q10 based on its capcity to scavenge free radical and to act as a bioenergetic agent [74],[75]. Patients who received the highest dose of Co-Q10 had significantly less deterioration in the Unified Parkinson’s Disease Rating Scale (UPDRS) than those in the placebo group [74]. Overall, this is a very promising area of PD treatment but much research still has to be done in the area of NTF therapy for PD.
Deep Brain Stimulation (DBS)
One other approach to PD treatment being used today involves surgically implanting electrodes into deep brain structures such as the globus pallidus, thalamus, and subthalamic nucleus [77]. High frequency stimulation of these structures may help normalize neuronal circuitry within the basal ganglia, and help resolve motor symptoms of PD [77]. Although deep brain stimulation has shown success in some patients more research is needed in this area.

Figure 10. Neuroprotective Pharmacological Interventions for PD [73]
Conclusion
Parkinson's disease is a neurological disorder characterized mainly by motor symptoms. The principal neurological abnormality that occurs is degeneration of dopamine-producing cells in the Substantia nigra pars compacta (SNpc) portion of the basal ganglia. PD progresses through five stages typically beginning with unilateral mild tremor and ending with complete physical dependence and death in an average of 13 years[4]. Treatments for the symptoms of Parkinson Disease include pharmaceuticals, physical and occupational therapy, and surgery. The mainstay pharmaceutical is Levodopa. Others include dopamine agonists, anticholinergics, amantadine, selegiline, and COMTs. So far, however, all of the PD treatments have undesirable side-effects and none stop the progression of the disease.
Research progresses utilizing the MPTP model in mice and primates. Most cases of PD arise sporadically, but newer etiological research is discovering a smaller number of cases caused by genetic mutations [3],[4]. Many of these mutations have been linked to mitochondrial dysfunction. Single gene mutations include a-synuclein (SNCA), LRRK 2, parkin (PRKN), DJ-1, PINK 1, and HTRA 2 genes[7],[2],[8],[1]. An area of research focus is neurotrophic factors like GDNF. Another promising treatment area, deep brain stimulation, remains effective for only a select few. Scientists will, however, continue to discover more about the causes, treatments, and ultimately, the cure for Parkinson's disease.
Return To Welcome Page
Go To PD Exercise
i
—
This looks good so far. I have noticed that you guys are going to be referencing your sources (which is needed) and there is a referencing style that is useful for wikidot that allows the site to build your bibliography numbers automatically for you so that you don't have to do it manually. Its pretty easy syntax tat you can find in the how to pages. I recommend that you start on this now and do it with each thing you load up on the page or it is going to be massively tedious and time consuming at the end.
Also, the introduction is good but it is based on a school text book. 99% of things that are loaded on this page needs to come from primary research. Text books are usually out of date by the time they are printed and don't offer the most recent evidence, even for something as basic as the intro. Try to find a good review article to make the intro from.
Otherwise looking good so far!
—
I know that this page is due soon and I wanted to give you some feedback regarding the content of your page. I really like the table of cellular components, it organizes it very well. However, the depth of knowledge is lacking. Just going by your list of references, you guys as a group need to go deeper into the pathways and cell bio of cellular components. One thing that I noticed that is a HUGE gap in your content is the lack of information regarding the inflammatory processes of PD. This is a MASSIVE component of the disease and I don't see anything about it. I don't know if this is because you haven't uploaded it yet or if its because it wasn't touched on. I know that there are tons of research out there on this w/human and animal models so I expected to see more content in this. Please rectify this before the due date or this might be reflected in your grade.
I think this page is well done and very easy to follow. I appreciated the flow between sections and summary statements.I liked your introduction and think that it set up the page nicely. I would however, remove the sentence about rehab and move that to your exercise page. Also, I'm not sure you should include the level of detail about clinical manifestations and disease progression on the cell bio page. Need to change "a-synuclein" to alpha or the alpha symbol.
In undergrad I was in a neuroscience course where we spent half the quarter learning about the cellular mechanisms of Parkinson's disease. So I have comments about some things that I was missing on your page.
First, I think you made a big omission regarding the animal models. After the MPTP drug was discovered to cause Parkinson's like symptoms the very first animal model for PD was created, which was rats. This is the classical animal model for this disease. Mice were not first as your page leads me to believe. The rats were treated with reserpine to model the disease.
I think your page would benefit from describing the D1 and D2 pathways in the presence and absence of dopamine, in its own section,not buried within a section and paragraph. You only touched on it, I think it should have more detail—which is important in understanding exercise effects and symptom presentation.
I was also missing neuromelanin and it's relationship with oxidative stress.
I didn't see overt mention of the Ubiquitin-proteosome system and it's relationship to Parkin—if you don't have Parkin you have a lot of misfolded proteins which is toxic to the cell.
I think it should also be mentioned that symptoms of PD don't appear until about 85% of DA is lost and the mechanisms underlying why this is—this is a big problem in the effective treatment and prevention of the disease. Some of the mechanisms include presynaptic compensatory mechanisms (such as pumping out more neurotransmitters-increased release of DA, increased turnover (breakdown), decreased DA reuptake (keep in the synapse for longer periods of time)) and post-synaptic compensatory mechanisms such as increasing the affinity of receptors for dopamine, increasing number of receptors and increasing the sensitivity of second messenger systems to dopamine. All these mechanisms contribute to why it takes 85% loss of DA before you see the first tremor.
Hope some of this is helpful for your page!
You may also want to look at medium spiny neuron and NMDA receptors involvement.
The overall organization and flow of the page is great. It is easy to understand the information presented. I only have a couple comments. In the inflammation section, near the end it has TGF-beta. Consider adding the β symbol instead of the word beta. Also, I agree with Cara that you may consider condensing the amount of clinical manifestation information about PD.
The overall organization and flow of the page is great. It is easy to understand the information presented. I only have a couple comments. In the inflammation section, near the end it has TGF-beta. Consider adding the [β] symbol instead of the word beta. Also, I agree with Cara that you may consider condensing the amount of clinical manifestation information about PD.
The page is nicely organized. I especially appreciate the breakdown of the cell bio components because it makes it easy to read and gives clear definitions of each. I found that to be difficult with several of our topics as many overlapped. So I think that's great! I'm not sure, but you may think about putting the interventions with the exercise page.
The page is nicely organized. I especially appreciate how the cell bio components are divided up, because it is easy to read and provides clear definitions of each. I know it was difficult to do that with several of our topics as many overlapped. So I think that's great! I'm not sure, but you may want to consider putting the interventions on the exercise page.