Introduction
Causes, Incidence, and Risk Factors
Spinal cord injury (SCI) is defined as any injury resulting from an insult to the spinal cord that disrupts its major functions, either completely or incompletely.[1] Spinal cord injuries can be caused by both traumatic and non-traumatic incidents that lead to damage to the spinal cord region.[2] The leading causes of traumatic spinal cord injuries include motor vehicle accidents (cars, motorcycles, bikes, ATVs), falls (leading cause for individuals over 65), sport related injuries (diving, skiing), industrial accidents, gunshot wounds, and assault (stab wounds).[2,3,4] Non-traumatic causes of spinal cord injury include stroke, infections, degenerative joint/bone diseases (RA, OA, osteoporosis, spinal stenosis), neoplasms, neurological conditions (ALS, MS), and medical/surgical complications.[2,5] Men, ages 15-35 years old, are most affected by SCI, but older individuals with degenerative joint/bone disease and those individuals prone to falls are also at a higher risk.[2]
Spinal cord incidence is difficult to measure because many people die either immediately upon injury or upon arrival at a hospital, and are not always included in incident reports.[6] However, the estimated annual global incidence of SCI is between 20 and 40 per million people.[3,6] Mortality, including both immediate death and death upon admission, is between 48 and 79 percent.[6] In the United States, around 11,000 new cases of SCI occur each year, with an estimated 250,000-400,000 total people living with SCI.[3,5] In terms of demographics, males are affected four times more than females, and the most affected age group is between 15 and 35 years old.[2] The majority of individuals with SCI are Caucasian (67%), with African-Americans (19%) and Hispanics (10%) being the next highest groups to be affected.[5]
Classification
Individuals with SCI are classified into groups based on the extent of paralysis and paresis they experience, and can be classified as either tetraplegics or paraplegics.[7,8] Tetraplegia (also called quadriplegia) results when there is partial or complete paralysis of all both the upper and lower extremities, as well as the trunk and pelvic organs, and is often the result of a cervical spinal cord lesion.[7,8] Paraplegia, which is often the result of a thoracic or lumbar spinal cord lesion, presents as partial or complete paralysis of the trunk, lower extremities, and pelvic organs, with upper extremities remaining intact.[7,8] When discussing the extent of impairments associated with SCI, neurological level of injuries and ASIA Impairment Scale (AIS) level are often discussed.[7] The neurological level of injury is the most caudal level of the cord that has both normal sensory and motor function.[7] When classifying the injury with the AIS, motor and sensory function is checked at the S4-S5 sacral segment to determine a complete or incomplete lesion.[7] In the case on a complete lesion (no sensation or motor function at S4-S5), there is often partial sparing of sensory and/or motor function in some of the segments below the lesion, and this is termed the Zone of Partial Preservation.[7] The different grades on the AIS (A-E) take into account the range of motor and sensory losses that can occur with incomplete and complete SCI.[7] Classifying injuries allows clinicians and researchers to effectively communicate the different clinical presentations of people with SCI.[7] Additionally, incomplete lesions can result in patterns of motor and sensory losses that have been grouped into several different clinical syndromes, as seen in Figure 2.[7] There is no clear-cut way to determine the degree of motor and sensory loss that will occur after a SCI, and the rest of this WIKI page will discuss the cellular mechanisms at-play after the injury that ultimately lead to the loss or preservation of motor and sensory function.
Prognosis
The most important factors predicting survival post-SCI include age, level of the injury, and neurological grade.[6] Individuals with a C1-C3 spinal cord lesion are about 6.5 times more likely to die, C4 or C5 levels are 2.5 times more likely to die, and C6-C8 are 1.5 times more likely to die than individuals with a thoracic, lumbar, or sacral lesion.[6] The AIS grade at initial injury can be predictive of prognosis. Individuals with an initial AIS grade C are most likely to progress toward a lesser severity grade (D,E) as compared to those with an initial grade of A or B.[7] Motor function in both upper and lower extremity muscles within the first month after injury can also be indicative of a return to normal motor function within the first year after injury.[7] Individuals with motor incomplete lesions who have normal quadricep strength within 2 months of injury have been found to have an excellent chance of becoming functional ambulators again.[9] On the other side of the spectrum, individuals who are initially motor complete with only light touch sensation intact are unlikely to return to ambulation.[9]
Financial Impact
The financial burden of living with SCI is enormous. On average, a person with an acute SCI will spend around 170 days in the hospital over the first two years post-injury.[6] The average initial hospital bill for SCI is around $95,000, with home modifications typically costing around $8,000.[6] After the initial hospitalization, rehabilitation, and recovery, the average person with a SCI will pay close to $8,000 a year on additional medical expenses, services, supplies, and adaptive equipment.[6] Home assistance and/or institutional care can add another $6,000 per year, on average.[6] Taking into account health care costs, inability to work, and additional costs from related complications, some individuals living with SCI end up costing society close to $200,000 per year and $1.5 million over a lifetime.[4,10] Apart from these measurable monetary costs, the person with a SCI still has emotional, physical, and social burdens to deal with.[3,6]
The huge financial impact of SCI on individuals, families, and society is cause for researchers and scientists to continue studying the cellular biology that impacts spinal cord injury. By better understanding the cellular mechanisms that contribute to the injury, better treatments can be continue to be developed in order to help alleviate some of this financial burden.
The Spinal Region Anatomy
In order to better understand the complex pathophysiological changes that occur following SCI, it is important to first understand the anatomy and function of the spinal cord region in a healthy individual. The spinal region is made up of neural structures housed within the vertebrae including the spinal cord, dorsal and ventral roots, spinal nerves, and meninges.[9] The spinal cord begins from the medulla oblongata, just above the C1 vertebral level and ends at the L1-L2 intervertebral space.[7,9] Inferior to this level, long lumbar and sacral nerve roots extend to form the cauda equina.[7,9]
The internal anatomy of the spinal cord is divided into white and gray matter, as seen in Figure 3 below.[7,9] The white matter contains the axons of both sensory and motor neurons that are ascending and descending along the spinal cord.[7,9] The white matter is further divided into dorsal, lateral, and anterior columns depending on the type of information being carried in the fibers running through each region.[7,9] The central part of the cord is made of gray matter which houses the terminal synapses of axons from other areas, neuronal cell bodies and dendrites, and glial cells (nonneuronal cells that help with spinal cord function and survival).[7,9] The gray matter is divided into dorsal, lateral, and ventral horns.[7,9] The dorsal horn contains mostly sensory neurons, the lateral horn (only present from T1-L1) contains the cell bodies of neurons involved with the sympathetic visceral innervation (help with autonomic regulation), and the ventral horn contains cell bodies of lower motor neurons that innervate skeletal muscle.[7,9]
Figure 1: Spinal Cord Anatomy

The spinal cord is surrounded on each side by ventral and dorsal roots, which contain axons carrying motor information to the periphery (ventral roots) and sensory information from the periphery to the cord (dorsal roots).[9] The cell bodies of the sensory neurons are housed in a structure called the dorsal root ganglion, which is outside of the spinal cord.[9] The cell bodies of the motor neurons however are housed in the cord itself in the gray matter.[9] The dorsal and ventral roots combine to form spinal nerves that exit via the intervertebral foramen and carry motor and sensory information to and from the periphery.[9]
The final component of the spinal cord anatomy are the meninges, which are layers of connective tissue that surround the cord.[9] Similar to the brain, the meninges are divided into pia mater, arachnoid mater, and the dura mater, which all serve to protect the spinal cord.[9]
Figure 2: Vascular Supply of the Spinal Cord

Vascular Supply
Blood is supplied to the spinal cord by three spinal arteries that run vertically along the cord.[7,9] The first, called the anterior spinal artery, runs along the anterior midline of the cord and supplies the anterior two-thirds of the cord, including both gray and white matter.[7,9] The second and third are called the posterior spinal arteries, and they run along either side of the posterior midline of the cord and are medial to the dorsal roots.[7,9] The two posterior spinal arteries supply blood to the posterior third of the cord.[9]. Figure 4 shows the normal distribution of blood flow to the spinal cord. These three spinal arteries use centrifugal and centripetal arterial systems to distribute blood to the entire cord.[7]
Centrifugal System
The anterior spinal artery divides into many small branches called sulcal arteries, which enter the anterior median fissure of the cord and supply the central cord region.[7] This area includes the majority of the gray matter and the inner half of the white matter.[7]
Centripetal System
The anterior spinal artery also gives rise to pial arteries, which combine with branches of the posterior spinal arteries and travel the outer circumference of the cord.[7] This system supplies the peripheral regions including the dorsal horns, most of the posterior white matter, and the outermost portion of the anterolateral white matter.[7]
Primary Injury
A spinal cord injury results in two mechanisms of injury, called the primary and secondary injury.[3,4,6] The impact of the direct mechanical trauma to the spinal cord causes the primary injury which sets off a cascade of widespread, progressive biochemical and cellular processes that make up the secondary injury.[3,4,6]
There are four main characteristic mechanisms of primary injury which include:[1,4,6]
- Impact plus persistent compression
- Impact alone with transient compression
- Acute distraction of the spinal cord
- Laceration or transection of the spinal cord
The most common mechanism of injury is the impact plus persistent compression, which can be seen following a burst fracture of the spine, a fracture-dislocation of the spine, or an acute disc rupture into the spinal cord.[1,4,6] Impact along with transient compression is often seen with hyperextension injuries in patients with an underlying degenerative spine disease.[1,4,6] Acute distraction of the spinal cord can occur following extreme flexion, extension, or rotation that causes shearing or stretching of the spinal cord.[1,4,6] Lacerations can occur from violent injuries or sharp bone fragment dislocations and vary from minor to complete transections.[1,4,6]
Following the initial trauma, neurogenic shock and systemic loss of autoregulation occurs.[1] The initial trauma causes local membrane damage, spinal cord compression, local vascular changes, increased glutamate release, edema, and inflammation which can ultimately lead to cell death.[1,3,7] The initial injury leads to both neuronal and endothelial cell membrane shearing, disruption of the vascular structures of the cord, and hemorrhaging.[1,3,4] There is greater damage to the gray matter than white matter due to the gray matter’s higher vascularity and metabolic demands.[1,3,4] The disruption in blood flow leads to local infarction caused by hypoxia and ischemia.[1] Additionally, the axons of neurons that span the injury site become physically damaged from the injury mechanism and the resultant micro-hemorrhages and edema perpetuates they cycles of neuronal damage and decreased nerve transmission.[1] In general it is thought the gray matter is irreversibly damaged within the first hour after injury, while white matter is irreversibly damaged within the first three days.[1]
Secondary Injury
Figure 3: Secondary Mechanisms of Injury

Secondary injury is a result of the progressive destruction from the primary injury that further exacerbates the injury to the cord itself. The main mechanisms which create the overall effects on the spinal cord are often associated with vascular changes, free radicals, excitotoxicity, and cell death.[3,6] Secondary injury often presents within two hours post-injury, where the damage begins to travel rostrally and caudally from the primary lesion.[3,4] At this point, edema and the hemorrhaging within the gray matter increase over the next few hours, before damage to the white matter becomes evident.[3,7] A brief synopsis of the different mechanisms involved with the primary and secondary injury can be seen in Figure 3.
Systemic Effects
Injury to the spinal cord can lead to neurogenic shock and respiratory failure through the disruption of the autonomic nervous system's (ANS) ability to regulate the body.[1] Neurogenic shock is a transient phase that occurs immediately after trauma in which spinal reflexes, voluntary motor and sensory function, and autonomic control are absent or depressed below the lesion.[7] The loss of autonomic regulation leads to the brief increase in heart rate and systemic blood pressure seen post-SCI, as well as the following prolonged bradycardia and hypotension.[1,6,11] The loss of tone in blood vessels leads to pooling of blood in the arteries and veins further from the heart causing the prolonged hypotension.[7] These initial changes lead to decreased peripheral vascular resistance, decreased cardiac output, hypoxia, and hyperthermia.[1,6]
Local Vascular Effects
Following the initial traumatic impact, local changes occur to the vascular supply of the spinal cord, ultimately leading to both hemorrhagic and ischemic states.[1,4,6,11] There is an initial reduction of blood flow, and within 15 minutes of the acute injury, hemorrhaging occurs in the gray matter and edema occurs in the surrounding white matter.[4,6,7,11] Edema in the cord can cause further compression of the meninges, leading to further disruption of the spinal cord by damaging its protective layers.[7] Vessels in the gray matter become swollen with erythrocytes causing small petechial hemorrhaging in the perivascular space.[1,4] The hemorrhaging extends rostrally and caudally over the next few hours following the initial insult.[1,4,7,11] Microcirculation, including venules and capillaries, experience more extensive damage than larger vessels.[1]
Ischemia begins to progressively worsen over the first few hours post-SCI, although the mechanisms are still unclear.[4,6,7] Possible causes include vasospasms which are induced from the initial trauma, intravascular thrombosis formation due to platelet aggregation, and the presence of increased norepinephrine in the blood supply.[1,4,6,7] Ischemia can lead to local edema and endothelial swelling and damage of capillaries and venules within 1-2 hours post-SCI.[6] The central gray matter and adjacent white matter are most affected acutely because the ratio of blood flow is approximately 3:1 for gray to white matter.[6] Axonal damage is also noted due to disruption of the myelin sheath.[4]
Obstruction of the anterior sulcal arteries and arterioles may lead to hemorrhagic necrosis at the primary injury site.[6,7,11] Overall the central nervous system is intolerant to ischemia and as little as 15-30 seconds without oxygen can lead to irreversible damage in neurons.[7] After the initial stage of reduced perfusion, a period of hyperemia occurs due to reduced perivascular pH from the accumulation of acidic metabolites (e.g. lactate).[1] This reperfusion can actually worsen the injury and increase necrosis because it produces harmful free radicals and other reactive oxygen species.[1]
One of the local vascular effects following SCI is infarction, or cell death due to loss of oxygen. Loss of oxygen in neurons perpetuates necrotic cell death in the CNS tissue through various mechanisms. The initial injury leads to loss of cell membrane integrity, vascular vasospasms, and thrombosis, which ultimately lead to a decrease in oxygen and glucose being provided to the cell.[6,4,11] Without sufficient oxygen and glucose, there is a disruption in the processes of glycolysis and oxidative phosphorylation, which are the main cellular cascades used to produce ATP in the cell. With a decrease in ATP, the cells experience disruption in their membrane permeability and changes in ion concentrations, release of lysosomal enzymes which break down the cell, and increases in intracellular calcium which sets of the process of calcium-mediated cell death.
Excitotoxicity
Excitotoxicity is defined as neuronal death caused by excessive or prolonged activation of glutamate receptors.[12] In uninjured spinal cords, glutamate is an excitatory neurotransmitter involved with the depolarization of axons of spinal neurons.[12] When an axon is depolarized, glutamate is released from the nerve terminals, where it then crosses the synaptic cleft to act on postsynaptic ionotropic receptors, including N-methyl-D-aspartate (NMDA), α-amino-3-hydrox-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate.[3,12] NMDA is a ligand-gated ion channel that permits entry of sodium and calcium ions and efflux of potassium ions.[12] AMPA and kainate are non-NMDA, ligand-gated channels that regulate sodium influx and potassium efflux when activated by glutamate.[12] NMDA receptors are involved in secondary glutamatergic synaptic transmission once the neuron has been depolarized by activation of AMPA/kainate receptors.[12] The activation of these ionotropic receptors leads to further depolarization; when a certain threshold is reached an action potential is generated.[12] Normally, glutamate is rapidly removed from the synapse through uptake systems including excitatory amino acid transporters (EAAT).[12] EAAT are proteins found in the membranes of glial cells (including astrocytes, oligodendrocytes, and microglia), neurons, and endothelial cells. They transport glutamate, along with other ions, across cell membranes, and quickly remove it from the extracellular space. This quick removal helps to maintain extracellular levels of glutamate and also stops the synaptic transmission. The glutamate loop in a healthy neuron can be seen in Figure 4.
Figure Number 4: Normal Glutatmate Loop

Mechanisms
Excitotoxicity occurs as a result of chronic depolarization of neurons caused by increased levels of extracellular glutamate.[1,12] Changes in sodium (Na+) and calcium (Ca2+) ion influx, as well as exocytosis of glutamate, all play a role in the mechanism of excitotoxicity.[12] Depolarization leads to increased activation of AMPA, and kainate.[12,13]. When glutamate binds to the AMPA or kainate receptors, depolarization occurs, which in turn activates voltage-dependent Na+ channels, causing even further depolarization and an increase in intracellular Na+.[3,12] Cells will begin to increase their influx of chloride ions to attempt to balance out the sodium influx.[3,12] Ultimately, the osmotic balance of the cell is so disrupted that the cell begins to increase its water intake.[3,12] Cell lysis occurs causing cellular contents, including glutamate, to leak back into extracellular space.[3,12] Additionally, there is increased activation of NMDA receptors, which leads to an increase in intracellular Ca2+.[12] Calcium ions enter the cell through the activated NMDA channels and further disrupt cellular homeostasis.[12] As more and more cell lysis occurs, glutamate levels increase, there is slowing of the glutamic acid transport chain, and calcium-mediated cell death begins.[12]
Cell death from excitotoxicity also occurs via changes in ion levels, calcium-dependent mechanisms, dysfunctioning mitochondria, and the production of free radicals and other reactive oxygen species (ROS).[12] Increases in Na+ influxes triggers an increase in Ca2+ influx through voltage-gated calcium channels, as well as reversal of sodium-calcium membrane exchangers.[14] High sodium levels also lead to edema and intracellular acidosis.[1] Sodium-potassium ATPase fails during excitotoxicity causing increasing accumulation of Na+, water, and Ca2+ into the cell, and increasing loss of potassium (K+) into extracellular space.[1]
Excitotoxic cell death also occurs in glial cells of the central nervous system (CNS). This form of excitotoxicity occurs via the glutamate receptors AMPA and kainate, which are found on oligodendrocytes (the myelin producing glial cells in the CNS).[3,14,15] AMPA and kainate have an increased permeability to calcium compared to neuronal cells. These oligodendrocytes have a poor ability to buffer calcium and thus are highly susceptible to excitotoxic cell death.[3] Extensive excitotoxicity can lead to ischemia, demyelination, axonal damage, inflammation, white matter atrophy, disruption of cell processes, loss of membrane integrity, and eventually cell death.[3,14,15]
Free Radicals and Reactive Oxygen Species
Glutamate neurotoxicity is also mediated by the generation of free radicals and reactive oxygen species.[12] Free radicals disrupt cellular organelles and initiate cell-programmed death, apoptosis.[12] NMDA receptor-mediated excitotoxicity initiates a cascade of events that ultimately leads to the genesis of reaction molecules that contribute to neuronal death through a variety of mechanisms.12] Lipid peroxidation can begin that can produce free radicals, as well as an alteration in the integrity of the sodium potassium pumps, which are described below.[12] Regular mitochondrial functions that produce free radicals can become damaging after SCI.
In the mitochondria, the free radical superoxide (O2-) is produced as a by-product of oxdative phosphorylation.[16] O2- is normally detoxified by superoxide dismutase (SOD) which produces the more stable moledcule of hydrogen peroxide (H2O2).[16] When transition metal ions such as iron are present, the hydrogen peroxide is reduced and a hydroxyl (OH-) free radical is produced.[16] Luckily, cells have built-in antioxidant systems to keep the levels of free radicals below a toxic threshold. One example is the mitochondrial antioxidant manganese superoxide dismutase (MnSOD) which detoxifies superoxide to produce hydrogen peroxide.[16] Another example is glutathione peroxidase (GPx) that can detoxify hydroxyl free radicals into water.[16] When free radicals are produced faster than they can be detoxified, or if the detoxification systems are impaired, the build-up of reactive oxygen species can lead to damage of mitochondrial proteins, lipids, and nucleic acids. With these impaired, oxidative phosphorylation cannot occur and more and more free radicals will be produced. Eventually this leads to apoptotic cell death.
Free radicals have been implicated as harmful contributors to the secondary injury following the initial SCI. Nitric oxide (NO) levels increase following SCI which impairs mitochondrial respiration by binding to respiratory enzyme Complex IV.[17] Superoxide anion (O2-)causes damage by impacting Complex III of the oxidative phosphorylation process.[17] A study by Wu et. al looked at the effects of these two free radicals on mitochondrial respiratory chain enzymes in male rats with complete thoracic SCI.[17] They found increased production of NO and increased levels of O2- in the spinal cord 3 days post-injury. The study also showed the two free radicals promoted apoptosis through different mechanisms.[17] NO led to the nuclear translocation of apoptosis inducing factor (AIF) by increasing activity of poly(ADP-ribose) polymerase-1 (PARP-1) and Bax.[17] O2- was responsible for mediating mitochondrial release of cytochrome c.[17]
Ion Channels & Concentrations
Potassium
Potassium (K+) channels are located on myelinated axons and facilitate in the production of action potentials in order to stimulate neuronal conduction.[9] They are also essential in cardiac muscle and assist in regulating hormone secretions in the body.[9] There are three types of K+ channels: 1) Fast K+ channels, 2) Slow K+ channels, 3) Na+-dependent K+ channel.[18] Although the majority of research following SCI focuses on the voltage-gated ion [fast] channels due to their prominent location on myelin, overexposure to any of these can further implicate secondary injury.[18] As discussed within various mechanisms of secondary injury, axonal damage after SCI is evident due to demyelination of the axons, leading to subsequent conduction failure.[3,6] Normally, opening of voltage-gated ion channels creates an influx of sodium (Na+) ions.[9] This rapid influx depolarizes the cell, forcing the Na+ channels to close, opening K+ channels and K+ is effluxed.[9] Demyelination exposes these K+ channels, increasing their activity which in turn shunts Na+, inhibiting action potential generation.[18] The inhibition interrupts signal conduction, which further damages cells and releases excitotoxins.[18]
Sodium
Sodium channels are found within neurons, myocytes, and glial cells.[9] Often membrane gradiences of other channels are dependent on Na+ ions.[9] Similar to K+ channels, Na+ channels are essential in generating action potentials and are affected by SCI.[9] Again, damage to the myelin sheaths disrupts the cellular homeostasis of Na+ and K+, consequently inhibiting action potential generation and creating axonal conduction dysfunction.[18] Also due to demyelination is activation of the Na+-Ca2+ exchanger, which increases intracellular levels of Ca2+, leading to a myriad of events that also induce secondary damage.[18,19] Disregulation of Na+ and K+ homeostasis has also been found to increase glutamate release which contributes to excitotoxicity post SCI.[18,28] Finally, because of their location on neurons, over-expression of Na+ channels has also been linked to neuronal excitability.[18] This correlates Na+ channel malfunction to central neuropathic pain, which can be present after injury to the CNS.[18]
Magnesium
Following SCI, there is a decrease in intracellular magnesium, which serves as a neuroprotector by blocking NMDA receptors.[1,20] Thus, decreased magnesium can contribute to the increase in intracellular calcium, and which indirectly contributes to calcium-mediated secondary injury.[1,20] Several studies have looked at the effects of magnesium therapy on acute SCI in rats and rabbits.[20,21,22,23,24,25,26,27] These studies have used weight-drop contusion and clip compression injury models to study the effects of administering magnesium sulfate at various doses (100, 300, 600 mg/kg) given immediately post-injury.[20,21,22,23,24,25,26,27] A dose of 600 mg/kg has been shown to be more effective than 300 or 100 mg/kg with regards to tissue sparing, reduction in apoptosis, lipid peroxidation, and function on the BBB Scale.[20] Most studies found success with administration within the first 30 minutes post-SCI.[20,21,22,23,24,25,26,27] However, additional studies have shown that magnesium administration may be given as late as 8 hours post-SCI and still show beneficial effects.[20] Although a dose of 600 mg/kg of magnesium sulfate has been shown to be optimal in mice, this does is outside tolerable human limits. Thus, for human treatment, lower doses of magnesium when combined with polyethylene glycol (PEG) have also been found to increase tissue paring at the primary injury site and improvements in BBB at 6 weeks post-injury.[20,21] With multiple ion concentrations being changed following SCI, it important to explore therapeutic means to regain homeostasis, and magnesium supplementation may be one way to do this.
Calcium-Mediated Secondary Injury
Another form of glutamate excitotoxicity occurs through the activation of Ca2+ systems. The continuous axonal depolarization from increased glutamate eventually leads to a massive increase in intracellular Ca2+ via voltage-dependent Ca2+ channels and opening of NMDA receptor channels.[12] Eventually, self-destructive calcium-dependent enzymes are activated which also trigger cell death.[12] Once intracellular levels of Ca2+ increase, nitric oxide synthase occurs, resulting in increased activation of poly (ADP ribose) polymerase-1 (PARP1).[16,29] PARP1 is an enzyme associated with cell differentiation and proliferation, and helps regulate DNA damage during natural occurring cell death.[29] Additionally, there is an increase in apoptosis inducing factor (AIF) translocation to the cell nucleus, damaging the DNA damage.[16,29]
Increases in intracellular Ca2+ levels leads to impaired cellular respiration, which is a function of the mitochondria.[1] Additionally, the increased Ca2+ levels stimulate calcium-dependent proteases (break down peptide bonds holding proteins together) and lipases (break down fats), including calpains, phospholipase A2, lipoxygenase, and cyclooxygenase.[1] The activation of lipase, lipoxygenase, and cyclooxygenase leads to the increased conversion of arachidonic acid into thromboxanes, prostaglandins, and leukotrienes.[1]
Catecholamines
Epinephrine (Epi) and norepinephrine (Nor) are catecholamines produced by chromaffin cells of the adrenal medulla located in the adrenal cortex. While Epi is specific to the adrenal medulla, Nor is also produced by postganglionic fibers of the sympathetic nervous system.[30] Catecholamine release is regulated by the sympathetic nervous system; when activated, increased concentrations of catecholamines are released into circulation.[30] The sympathetic nervous system is a part of the autonomic nervous system which is controlled by sympathetic reflex circuits and modulation by higher brain centers.[31] Under normal circumstances, stress is a primary stimulant for the release of catecholamine into the blood stream.[30]
The sympathetic nervous system has a powerful influence on both cardiovascular and pulmonary functions, as well as metabolic processes.[31] Spinal cord injury results in increased activation of the sympathetic nervous system (SNS), causing increases in plasma concentrations of the catecholamines epinephrine and norepinephrine (NE).[31] The alterations in catecholamine levels correspond to changes in heart rate, stroke volume, blood pressure, and metabolism found post-SCI.[31]
Within 30 minutes of spinal cord injury, NE levels double and continues to increase throughout the first hour to four times the original level.[32] A steady decrease occurs over the next few hours until baseline values are reached, typically around four hours post-injury.[32] Increases in NE levels have been linked to increased hemorrhage and necrosis levels in the central gray matter of the spinal cord.[32] These levels of NE are toxic and can induce vasospasms of the vascular system which further decreases spinal cord blood perfusion and leads to cell necrosis and vascular rupture.[7,32]
Catecholamines have been found to possess strong influences on the immune system. This relationship is made possible due to the presence of β & a-adrenergic receptors found on the plasma membranes of neutrophils, macrophages, T and B lymphocytes and natural killer cells.[33] Also, noradrenergic nerves have been found to innervate immune cell storage tissues, thus stimulating their release into circulation.[34] Therefore, catecholamine release corresponds to a large increase in leukocytes and lymphocytes. While neutrophils numbers will increase, catecholamines have also been found to inhibit their functions through decreasing their ability to phagocytose foreign material or release lysosomal enzymes.[34] Catecholamines also inhibit T helper cell’s release of antigens through binding to β2-adrenoreceptors on the cell membranes.[35] Catecholamines have also been shown to inhibit pro-inflammatory cytokines such as TNF-α, IL-1β and IL-12 from dendritic cells, monocytes and blood cells, while also facilitating the production of the anti-inflammatory IL-10 cytokine. [35] Catecholamines have been found to lead to an overall decrease of immune cells after prolonged or repeated exposure, specifically NK and lymphocytes.[36, 35,37]
Cortisol
Cortisol is the primary glucocorticoid hormone released from the adrenal cortex whose functions include helping with fat, protein and carbohydrate metabolism, increasing blood glucose, and suppressing the immune system. Cortisol release is stimulated by adrenocorticotropic hormone (ACTH) released in circulation from the pituitary gland in response to physical or mental stressors.[38] ACTH release is stimulated by corticotrophin-releasing hormone (CRH) from the hypothalamus.[38] Following spinal cord injury, circulating cortisol levels can remain high for several months.[37] It is through a negative feedback loop that high levels of cortisol will suppress itself through inhibition of CRH and ACTH release.[39] Individuals with spinal cord injuries, both paraplegics and tetraplegics, were found to have relatively higher levels of cortisol as compared to non-injured age and gender matched controls.[40] Cortisol inhibits many pro-inflammatory mediators such as cytokines (IL-1 , 2, 3,4,6,and 8, TNF-α,and IFN-γ), phospholipids, and proteases.[41] Other anti-inflammatory actions of cortisol include promoting antigen uptake by phagocytes, decreasing proliferation and increasing apoptosis in eosinophils and T-lymphocytes.[35] Through these interactions, it becomes apparent cortisol leads to an overall decrease in immune system function.
Apoptosis and Necrosis
Secondary injury consists of two mechanisms of cell death which assist in mediating further spreading of the injury.[3,6] The first being necrosis, followed by apoptosis. Necrosis is often described as premature cell death amongst living tissue.[6] It is often characterized as swelling of the cell, resulting in interruptions in the cell’s homeostasis, leading to cell lysis.[6,19] This event is caused by external factors, such as infection or trauma. The second mechanism, apoptosis (often referred to as programmed cell death), is a natural occurring cell death within the body.[3,19,42] This is a domino-like effect of “cell suicide” in an organized, sequential fashion involving a myriad of various biological events.[3,19,42] Although the two are in contrast to one another, necrosis and apoptosis occur simultaneously during the early stage of secondary injury. In later stages, apoptosis is present independently of necrosis.[3,19,42]
Necrosis kills cells via cell lysis by disrupting its homeostasis, resulting in swelling and later death of the cell.[6,19] This then causes the extracellular environment to be exposed to the intracellular content, creating an inflammatory response.[6] However, there has been no evidence of new proteins present in this process.[6] Necrosis is an immediate component of secondary injury, and travels approximately two vertebral segments rostrally and caudally from the initial lesion.[3] Necrosis affects the immune system differently than apoptosis.[6] Instead of signaling an immune response, necrosis actually alters the signal to the system, in an attempt to prevent phagocytes from recognizing them.[6]
Apoptosis is a natural occurring process that plays an important role in tissue development by balancing cell death and cell division.[19] Following SCI, apoptosis occurs among astrocytes within the epicenter, and in microglia in the surrounding gray and white matter.[42] Traumatic SCI sets off mechanisms of apoptosis which contribute to other detrimental effects in the body, such as neurodegeneration, ischemia, and inflammation.[19] Following SCI, three apoptotic mechanisms have been identified which use both intrinsic and extrinsic pathways to mediate apoptosis[43]:
- Ligand binding death receptor
- Mitochondrial-mediated
- Endoplasmic reticulum (ER) apoptosis
Extrinsic Pathway
The extrinsic pathway occurs at the cellular membrane and is carried out by ligating TNF related death receptors (TNF to Fas) on cell plasma membranes.[19,44] Once these death receptors are activated adapter proteins are attracted to the binding site.[3,19,44] Adapter proteins initiate a death-inducing signaling complex (DISC) creating conformational changes within the cell.[44] DISC works by activating procaspase-8, which in turn activates caspase-3 and pro-apoptotic protein Bid.[3,44] Cystolic Bid translocates to the mitochondrial membrane, promoting Bax and Bak binding to the membrane and altering its permeability.[44] This results in mitochondrial dysfunction and is the starting point of the intrinsic pathway.[3,19,44,45] Figures 5 and 6 show both the intrinsic and extrinsic apoptotic pathways.
Intrinsic Pathway
The mitochondria is the prominent organelle involved with the intrinsic pathway, and is often controlled by pro- and anti-apoptotic proteins.[19] These proteins are described in Table 1. Alterations in the mitochondrial membrane permeability allow cytochrome c to be released from the mitochondria. Cytochrome c is a protein stored in mitochondria that participate in the electron transport chain (ETC).[3,19,42] When expelled into the cytosol, chromosome c then binds with apoptotic protease-activating factor-1 (Apaf-1) and procaspase 9, forming cytosolic apoptosome, a protein that further induces apoptosis.[19,44] This creates a trickling effect which ultimately activates caspases (-3, -6, -7) and cysteine proteins which are designed to induce apoptosis.[3,6]
This particular process requires energy,[6] and often involves the mitochondria, which are initially spared along with the overall cell integrity,[3,6,19] demonstrating the mitochondrial-mediated mechanism mentioned earlier. Initially, no inflammatory response is noted, nor are the contents of the cell released as a toxin in the extracellular environment.[6,42] Although exact mechanisms are somewhat unclear, apoptosis is regulated by a myriad of cell signals. Intracellular signals occur following a stress to the cell, which consequently leads to cell suicide. These apoptotic signals result in a protein, cytochrome c, to be released from the cell via intrinsic and extrinsic pathways. This in turn activates an enzymatic family of cysteine proteases, called caspases.[3,19] Caspases, specifically caspase-3, destroy the cell through fragmentation of DNA and proteins, preventing interactions with other cells and DNA replication.[3,19] These events breakdown the cell and induce signals which activate phagocytosis.[3,19]
As previously stated, two stages of apoptosis have been identified during the secondary injury of SCI. The initial stage occurs in addition to necrosis at the lesion site and is present approximately 6 hours post-injury and remains for a few days following the injury.[3] During this stage, the necrosis and apoptosis work simultaneously in degenerating multiple cell types.[3] The second, later stage occurs approximately one week post-injury, once apoptosis presents at areas away from the central lesion.[3] It is specifically located in the white matter of the spinal cord, affecting oligodendrocytes and microglia, and incorporates components of both the intrinsic and extrinsic pathways.[3,6] As previously mentioned, oligodendrocytes provide myelination to axons in the nervous system.[9] When apoptosis begins in the axons, it results in demyelination of the tracts spared following primary injury, and can hinder functional recovery in patients with SCI.[3] Death of oligodendrocytes has been identified in ascending and descending white matter tracts, around the lesion epicenter and in areas of Wallerian degeneration.[42] It has been suggested that microglial activation may result in death of oligodendrocytes, and is most prominent at approximately one week post-injury.[6] As a consequence to SCI, death of these cells is often associated with axonal death, interrupting the cell signal pathways resulting in the motor and sensory deficits present following SCI.[19] The delayed demyelination is somewhat advantageous, allowing a small window of opportunity to provide treatment to these patients which is discussed later.[3]
Another form of apoptosis occurs under the control of AIF.[29,44] This is a mitochondrial protein that is independent of apoptotic pathways that utilize the caspases.[29] Normally, AIF acts as a cell protector against oxidative stress.[29] However, once cell death is initiated following SCI through various mechanisms (excitotoxicity, ischemia, etc.), AIF is released from the mitochondria at the same time as cytochrome c.[29,44] AIF translocates to the cell nucleus at this time, and also defragments DNA.[29] This mechanism of AIF cell death is the key player in apoptosis when the caspases are inhibited.[29] As mentioned in earlier, AIF has also been linked to cell death through excitotoxicity when the NMDA receptors increase Ca2+ influx. This is explained earlier under excitotoxicity.
Mitochondrial Impact
For a brief review on the function of healthy mitochondria, please click here.
Mitochondria are often referred to as the “powerhouse” of the cells.[19] They are the primary energy source for the cell, generating adenosine triphosphate (ATP).[19,46] They are also responsible for cell differentiation, signaling, and cell survival or death.[19,46,47] Mitochondria consist of an inner and an outer membrane (IMM and OMM, respectively), and works with the endoplasmic reticulum (ER) to regulate intracellular calcium (Ca2+) homeostasis.[19,46] Mitochondria have their own DNA (mtDNA) and house various proteins that participate in the aforementioned roles.[19] Fission and fusion of the IMM and OMM is crucial in maintaining a steady state within the mitochondrial and controlling the natural process of apoptosis.[19] The OMM surrounds the IMM, with the intermembrane between the two.[19] This intermembrane contains a protein-rich matrix that holds mtDNA.[19] Mitochondrial fragmentation can occur if these membranes become unbalanced.[19,46,47] The Bcl-2 family of proteins regulate the permeability of the mitochondrial membranes.[3] They control the release of cytochrome c from mitochondria and consist of pro- (Bax, Bak, Bid, Bad) and anti-apoptotic groups (Bcl-2, Bcl-xL).[3]
Fusion occurs when the OMM and IMM become joined together.[19] This process is regulated by GTPases mitofusions (Mfn1 and Mfn2).[19] If fusion is interrupted, there is an increase in mitochondrial fragmentation, inducing apoptosis.[19] Fission is the separation of the membranes. If it increases, mitochondrial fragmentation also increases.[19] Fission is regulated by proteins of mitochondrial fission 1 protein (Fis1), dynamin related protein (Drp1), and endophilin B1 (Bif1).[19]
Much research has been done studying mitochondrial dysfunction following SCI. As previously described, excitotoxicity occurs in consequence to neuronal injury post-SCI.[46,47] Research has linked excitotoxicity to increased buffering of Ca2+ by the mitochondria, leading to mitochondrial dysfunction and interrupting the electron transport chain (ETC).[46,47] The ETC is a process induced by the mitochondria as a means of providing energy to the cell.[47] Here, the mitochondria produces ATP and prematurely reduces oxygen, forming reactive oxygen species (ROS).[47] Disruptions in this process can alter the production of ATP and ROS, consequently increasing production of ROS and negatively impact mitochondrial homeostasis.[46,47] These mechanisms take place within the first 24 hours following SCI.[46] The increased Ca2+ uptake by the mitochondria is also correlated with increased membrane permeability.[19,46,47] Increased membrane permeability opens pores within the membranes referred to as mitochondrial permeability transition pore (mPTP), and leads to disruptions in the electron transport system, decreased ability to produce ATP, and increased production of ROS.[19,46,47] Swelling of the mitochondria occurs, causing a collapse of the membrane and inducing the release of cytochrome c leading to irreversible apoptosis.[19,46,47]
Several factors can contribute to secondary injury following SCI, many of which include functions of the mitochondria. Because they have such an imperative role in cell survival, any disruption to its integrity or processes can have a detrimental effect on the deficits the patient may sustain and the his/her overall recovery.
Table 2: Proteins Involved in Apoptosis[19]
| Pro-Apoptosis | Bax | Induces apoptosis via binding to and increasing permeability of OMM, releasing cytochrome c |
| Pro-Apoptosis | Bak | Induces apoptosis via binding to and increasing permeability of OMM, releasing cytochrome c |
| Pro-Apoptosis | Bid | Inserts Bax onto OMM; Refer to Bax function |
| Pro-Apoptosis | Bim | Facilitates apoptosis induction; Exact mechanism unidentified |
| Anti-Apoptosis | Bcl-2 | Inhibits apoptosis; Regulates OMM permeability with Bax and Bak |
| Anti-Apoptosis | Bcl-xL | Inhibits apoptosis; Regulates OMM permeability with Bax and Bak |
Heat Shock Proteins
Heat shock proteins (HSP) are protective proteins which are induced in response to a stress.[48,49] These proteins protect against injury, facilitate recovery, and participate in a defensive mechanism against subsequent stressors.[48,49] When proteins become injured, the folding of that particular protein becomes misshaped, disrupting its function. HSPs augment the refolding of those proteins in order to restore function and promote recovery in injured species.[48,49] Two forms of HSP are highly associated with apoptosis following SCI: HSP-27 and -70, labeled based on their overall shape.[48,49] When apoptosis is initiated by stress-induced factors, HSPs step in to inhibit this process.[3,48,49] HSP-27 is involved in the Fas-ligand binding mechanism of apoptosis. [48,49] Within this apoptotic-mediated mechanism, HSP-27 prevents the ligand binding on the cell membrane, preventing apoptosis. Specifically, HSP-70 has been investigated by Beere et. al.,[49] who recognized its ability to prevent the caspase activation via cytochrome c. HSP-70 prevents apoptosome formation by binding to Apaf-1, reducing its ability to recruit procaspase-9, thus inhibiting apoptosis.
Figures 5 & 6: Apoptosis and Mitochondria (Intrinsic and Extrinsic Pathways)


microRNA
During the natural occurring process of apoptosis, molecules known as microRNA (miRNA) have been identified as key regulators to maintain a homeostatic state in healthy individuals.[50] miRNA are considered a shorter chain (~22) of ribonucleic acids (RNA) and help regulate gene expression in the neural system.[50] Essentially they silence genes that have, or are, undergoing transcription through messenger RNA (mRNA), preventing the capability of the cells to carry out their roles.[50] Hundreds of these miRNAs have been identified, and are typically named by the order in which they were discovered.[50] Of these, several have been identified to interrupt transcription of various cellular components identified in apoptotic pathways.[50] Naturally, because of the implications apoptosis has following SCI, some research has recently been geared toward studying the involvement of miRNA.[50] Studies have shown that an insult to the spinal cord impacts miRNA, but the effects are dependent on the roles of each. One study[50], observed alterations in miRNA following contusion to the thoracic spine in Sprague-Dawley rats. They looked at the effects at 4 hours post-SCI, as well as 1 and 7 days post-SCI.[50] Two hundred and sixty-nine miRNAs were identified in the spinal cord, and approximately 60 were altered following SCI. Of these 60, some have been identified as dominant players in the regulation of apoptosis (Table 1).[50]
Table 1: Roles of miRNAs in Healthy Models[50]
| miRNA | Role of miRNA |
|---|---|
| miR-Let-7a | Pro-apoptosis: Acts on upstream cellular components |
| miR15b | Pro-apoptosis: Inhibits Bcl-2 |
| miR16 | Pro-apoptosis: Inhibits Bcl-2 |
| miR21 | Anti-apoptosis: Inhibits PDCD4 and/or PTEN |
PDCD4: Programmed Cell Death 4 - Pro-apoptosis; Activates caspases
PTEN: Phophatase and tensin homolog - Pro-apoptosis; Acts upstream of apoptosis by activating Bad
Inhibitors of Apoptosis
Inhibitors of apoptosis (IAP) are anti-apoptotic proteins that regulate apoptosis by protecting the cell from death inducing signals.[44] They consist of ~70 amino acid domains, termed baculoviral inhibitory repeat (BIR) which have been found to assist in the regulatory process of apoptosis.[43] Several have been discovered, but only a few (XIAP, cIAP1, cIAP2) will be discussed here as key players in apoptosis following SCI.[43]
X-linked Inhibitor of Apoptosis
The x-linked inhibitor of apoptosis (XIAP) is present in most tissues throughout the human body, with the exception of blood leukocytes.[44] Specific to the spinal cord, XIAP is located within the white matter.[43] It has been found to regulate apoptosis by inhibiting caspase-3 and -7 and binding to caspase-9 once apoptosis is initiated following SCI.[44,43] Immediately after an insult to the spinal cord, XIAP is damaged, interfering with its ability to regulate apoptosis, thus actually inducing apoptosis.[43] When this occurs, other IAPs (cIAP1 and cIAP2) tend to increase in number to compensate for the loss of XIAP.[44] Interestingly, this compensation is not recognized until approximately one week following SCI.[44] This seemingly creates a window for apoptosis to cause more damage during secondary injury.
Smac/DIABLO
When the spinal cord undergoes apoptosis following injury, the mitochondria actually release two chemicals which act on caspases to further augment apoptosis.[44] In addition to cytochrome c as discussed earlier, a second mitochondrial activator of caspases (Smac) is also released in humans.[44] If referring to mice however, this same caspase activator is known as direct IAP binding protein with low pI, or DIABLO.[44] This chemical binds to XIAP, dysregulates it, and inhibits XIAP’s ability to bind to the caspases.[44,43] Specifically, Smac/DIABLO actually breaks down the already binded XIAP and caspase.[44] The end result of this is a greater degree of apoptosis, and further damage to the spinal cord during secondary injury.
Ligand-binding death receptor
The XIAP is involved in the ligand-binding death receptor mechanism involving the TNF receptor and Fas ligand.[44] Here, XIAP breaks down, separating BIR1 and BIR2 domains from BIR3, allowing BIR1 and 2 to inhibit to caspase-3 and -7.[43] The mechanism by which BIR1 induces apoptosis is somewhat unclear, but some suggest that it may be a binding site for other IAPs.[44] The BIR2 domain is actually highly attracted to caspase-3 and binds directly to it, preventing other substrates from binding, thus inhibiting apoptosis.[44,43]
PGC-1α
Peroxisome proliferator-activated receptor γ co-activator 1 α(PGC-1α) belongs to a family of nuclear hormone receptors that serve as ligand-gated transcription factors for cells.[51,52] PGC-1α has been studied for its key role in regulating the gene transcription for mitochondrial biogenesis in the liver, heart, and skeletal muscle, and more recently looked at for its role in brain tissue and neurodegenerative diseases.[51,52,53]
Mitochondria biogenesis is the process by which a cell makes new and/or more efficient mitochondria, and can be stimulated by different signaling pathways during cellular exposure to oxidative stress or environmental stimuli such as exercise.[53,54,55] These new mitochondria can be produced from the transcription of genes from either the cell’s nuclear DNA or from the mitochondrial DNA (mtDNA).[54,55] As a result of mitochondrial biogenesis, there is greater enzyme production to stimulate the oxidative functions vital to a cell’s survival.[54,55] One of the main regulators of this transcription process leading to mitochondria biogenesis is peroxisome proliferator-activated receptor γ co-activator 1 α(PGC-1α).[51,52,54,55] PGC-1α is a co-activator of nuclear respiratory factor 2 (NRF-2), and together these molecules co-activate nuclear respiratory factor 1 (NRF-1).[51,52,54,55] NRF-1,2 upregulation leads to increased activation of mitochondrial transcription factor A (Tfam) which is responsible for regulating the level of mtDNA content and gene expression.[51,52,54,55]
As described above, SCI has a huge impact on mitochondria death, leading to impaired cell function and ultimately apoptosis. PGC-1α and the other molecules involved with its signaling pathways could serve as a potential means to create new mitochondria and thus re-establish the energy supply needed to maintain cell functions. Endurance exercise has been shown to increase the expression of PGC-1α, NRF-2, NRF-1, and Tfam in skeletal muscle ultimately leading to improvements in mitochondrial biogenesis, and these implications will be further described on the SCI Exercise Page.[53,54,55,65]
An additional implication for the potential effect of PGC-1α in SCI comes from the link between mitochondria and oxidative stress. Following SCI, the cells in the spinal cord are subjected to various reactive oxygen and nitrogen species, as detailed above, and this leads to the production of endogenous antioxidants to try and detoxify the cellular environment.[51] However, the oxidant/antioxidant balance is disturbed after SCI which eventually leads to accumulation of free radicals that can trigger mitochondrial-induced apoptosis.[51]
Animal studies have examined the role of impaired mitochondrial function and decreased expression of the genes that are involved with the cellular respiration process of oxidative phosphorylation in diseases such as Parkinson’s disease, Alzheimer’s Disease, and Huntington’s Disease.[51] In these studies, PGC-1α-knockout mice showed lesions (from cell death) in areas of the brain that have been known to affect individuals with these disease.[51] This implies that therapies aimed at increased PGC-1α could be a treatment in these neurodegenerative diseases that involve both mitochondrial dysfunction and damage from oxidative stress.[51] While SCI was not directly examined in these studies, the oxidative stress, mitochondrial impact, and apoptosis sections detailed above clearly show both these processes are at play in the pathology of SCI. Additional proof that PGC-1α could play a role in SCI stems from its regulation of endogenous antioxidants found in neurons such as glutathione peroxidase, catalase, superoxide dismutase 2 (SOD2) and uncoupling protein 2 (UCP2).[51] SOD2 and UPC2 are proteins produced by the mitochondria in the face of oxidative stress, thus providing a key link between PGC-1α, mitochondria, and oxidative stress, the latter of which have been implicated in SCI cellular pathology.[51] Studies examining the effects of PGC-1α on SCI are almost non-existent, but by applying the role of PGC-1α in regulating mitochondrial function and oxidative stress, it is clear that further research should be done.
Figure 7: Upregulation of ICAM and VCAM: Peripheral Cell Adhesion

Immune system
For a review of the major cells involved with the immune system response, please click here.
The CNS is considered “immuno-privileged” through isolation from the circulatory system by the blood brain barrier (BBB). The blood brain barrier is composed of a series of tight junctions between endothelial cells which limits the passage of circulatory components, including peripheral immune cells. Following spinal cord insult, peripheral immune cells are recruited to the injured site by local epithelial tissues and macroglia that release mediators to initiate the acute inflammatory response.[59] Pro-inflammatory mediators include such cytokines as: tumor necrosis factor α (TNFα), interleukin (IL)-1, IL-6 and IL-8 which are released to break down extracellular components, increases vascular permeability, and signals for leukocyte/lymphocyte recruitment to the site of injury.[3,66] Peripheral immune cells are transported across the BBB by the adhesion molecules intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) being enhanced on the epithelial and other cells, allowing leukocyte and lymphocyte adhesion via a receptor-ligand pair with lymphocyte function-associated molecule-1 (LFA-1).[66,68,69] See a and b of Figure 7.[70] The integrity of the BBB is further impaired through degradation of extracellular matrix and proteins by mediators such as matrix metalloproteinases (MMPs).
Innate Immune System
Innate immune cells include resident microglia/macrophages, recruited monocytes (neutrophils and macrophages), and dendritic cells.[59] Once activated, these cells perform many functions that include phagocytosis of cellular debris, destruction of foreign bodies, and acting as antigen producing cells (APCs) when their toll-like surface receptors bind to pathogens.[63] These activated APCs present antigens/peptides taken from phagosome digestion of foreign cells to T-lymphocytes using major histocompatibility complex II (MHC II) molecules embedded in their membrane.[63] Binding to pathogenic components leads to the production of pro-inflammatory cytokines by innate immune cells, further exacerbating the inflammatory reaction.[63]
Neutrophils
Depending on the mechanism of injury, various circulating monocytes may be immediately present at the site of injury, such in the case of an injury involving direct disruption of the BBB, leading to a tear which allows blood infiltration into the spinal cord injury site. Human IL-8 is a pro-inflammatory chemokine responsible for activation of neutrophils and their transport to the location of injury, making them among the first haematogenously derived monocyte to arrive at the injury site.[3] Neutrophils first appear attached to venules within 6-12 hours following injury, peaking at 24 hours in the CNS, and begin declining at 48 hours.[3,72] Though present a relatively short amount of time, they have a huge impact in the injured site. Neutrophils primary function is cleaning the injured area, through phagocytosis of debris, waste, and foreign bodies that may impair healing or cause infection. Neutrophils also act as antigen producing cells (APCs) in the presence of pro-inflammatory antigens such as TNFα and interleukins.[73] Their exposure to pro-inflammatory mediators facilitate the generation of oxygen-derived reactive intermediates, production of proteases, and the synthesis of potent mediators such as leukotriene β4, MMPs, and platelet-activating factor (PAF) which further increase vascular permeability and signaling for leukocyte recruitment.[73] Additionally, neutrophils have been found to increase the inflammatory and immune response through the up regulation of cytokines such as interleukin-1β (IL-1β), IL-1 and TNFα, as well as the chemokines IL-8, macrophage inflammatory protein-1 a (MIP-1α), and MIP-1β.[3,73] Conversely, neutrophils also excrete anti-inflammatory cytokines such as receptor antagonist (IL-1ra) to limit the injurious effects of excessive and sustained inflammation.[74] Neutrophils are largely replaced by macrophages by 48 hours, however they can be found in small quantities up to 10 days following CNS injury.[72,75]
Macrophages
Microglia are the native macrophage-like immune cells of the CNS which are dormant until injury or infection stimulates the cells to produce antigens and induce inflammation.[76] Because microglia are the local immune cells, already present at the time of injury, they have been shown to be activated in as early as one hour.[58] While some haematogenously derived macrophages may be present within minutes following injury due to disruption of the BBB, most arrive through chemotaxis within 2-3 days.[3,59] Macrophages, like neutrophils engulf foreign debris and apoptotic waste; however it has been demonstrated they remain at the injury site much longer. Studies of macrophage populations in rats show they do not start declining until 7 days post injury.[60] While their numbers are declining, activated microglia and lower populations of macrophages remain present at the injury for weeks to months.[75] Microglia and hematogenously derived macrophages also release pro-inflammatory cytokines such as TNFα, IL-1 and IL-6.[3] IL-1β at the protein level released by microglia converted macrophages has been discovered within five hours after spinal cord injury in humans.[59]
Adaptive Immune System
B and T-Cell Lymphocytes
Lymphocytes are antigen specific immune cells that circulate through the bodies’ lymphatic system. Following spinal cord trauma, they are attracted to the injury site through antigen presentation and the recognition of cytokines and chemokines released by injured microglia, endothelial cells, and activated neutrophils.[130] When activated, many B-lymphocytes produce antibodies while some are responsible for memory in adaptive immunity. Antibodies are used to mark antigens to facilitate their phagocytosis and further activate the immune cells by cross linking specialized antibody Fc receptors.[61] B-cell populations peak at 7 days and are decreased between one and two weeks post-injury as antigens are removed and endogenous regulatory cascades suppress the response.[61,62]
T-lymphocytes persist at the injury long after B-cell numbers decline, up to 1-2 months.[63] T-cells are classified as helper cells, CD4+ or cytotoxic cells, CD8+ depending on their phenotype and function. CD4+ cell become activated when they are exposed to peptide antigens by MHC class II molecules, which are expressed on the surface of antigen presenting cells (APCs). They proliferate, creating large numbers of daughter cells specific for the recognized antigen. Lymphocyte clone cells either become memory cells and are stored, or effector cells that function in the immune response. Effector cells are involved in orchestration of the immune response, and release cytokines that are toxic to target cells.[62] Pro-inflammatory cytokines released by CD4+ cells include interferon-gamma (IFN-γ), TNF-α, and IL-6. Additionally, CD4+ cells release cytokines with anti-inflammatory properties such as IL-4, IL-10 and transforming growth factor-beta (TGF-β).[63]
Figure 8: The Glial Scar

Glial Scar
As the primary and secondary reactions to the trauma subside, the necrotic area of the spinal cord is gradually resorbed and replaced by scar tissue, cysts, or cavities.[7] The formation of glial scars help to clean up debris and block off the primary injury site.[3] The acute injury site is filled with debris from myelin and oligodendrocytes (myelin-producing glial cells), which is inhibitory to axonal regrowth.[3] Within the first 48 hours, microglia migrate toward the injury site where they phagocytose the myelin debris, which can help promote axonal sprouting.[3] Next, the glial scar is infiltrated by oligodendrocyte-precursors that include NG2 and platelet-derived growth factor-α (PDGFR-α).[3] In unmyelinated areas, these pre-cursors are able to mature and differentiate into new oligodendrocytes.[3] Meningeal cells also migrate to the glial scar and re-form disrupted cells by continuing to wall-off the primary lesion.[3] Finally, astrocytes migrate to the glial scar where they increase their production of a protein called glial fibrillary acidic protein (GFAP).[3] GFAP helps with astrocyte-neuron communication and also with further formation of the glial scar.[3] Astrocytes then envelop the necrotic primary injury site with a glial lining and fill up the empty space of the lesion.[3] In order to re-establish and maintain the integrity of the lesioned area, the astrocytes secrete trophic factors and cytokines to help regulate the blood spinal cord barrier, excitatory amino acid concentrations, and ionic concentrations.[3] Fibroblasts and Schwann cells also migrate toward the glial scar to further help with remyelination.[3]
Matrix Metalloproteinases (MMPs)
Overview of Function
Matrix metalloproteinases (MMPs) belong to a family of zinc- and calcium-dependent extracellular endopeptidases that degrade the extracellular matrix and other extracellular proteins.[11,129,129,130,130,131] At least 23 MMPs have been identified in mammals and they are divided into groups based on their protein structure and substrate specificity.[130,130] Two MMPs that are widely implicated in SCI, MMP-2 and MMP-9, are classified as gelatinases.[130] MMPs can be regulated by the production of endogenous inhibitors of MMPs (TIMPs) and reactive oxygen species, which are increased in response to SCI.[130,131] In general, MMPs control a wide range of signal transduction pathways that impact cell differentiation, transformation, cell migration, regulation of growth factor activity, cell survival or apoptosis, angiogensis, and cell signaling.[129,130] MMPs execute this through the breakdown of extracellular and membrane-bound adhesion and structural molecules, proteoglycans, cytokines, and trophic factor ligands and receptors.[129]
MMPs have been implicated in spinal cord injury pathology and have also become a huge target for therapeutics.[11,129,129,130,130,131] MMPs are essential for remodeling the extracellular matrix, tissue morphogenesis, and wound healing.[71,130] In the spinal cord, MMPs can degrade components of the basal lamina (especially occluding and claudin), leading to disruption of the blood-spinal-cord barrier, increased production of reactive oxygen species, increased demyelination, increased immune system response.[11,129,130,131]
MMP9
MMP9 is expressed by inflammatory cells including macrophages, lymphocytes, neutrophils, and endothial cells.[129] MMP-9 has the ability to degrade gelatin, collagen, and elastin, as well as the basic protein unit that makes up myelin surrounding axons in the gray matter of the SC.[129] MMP9 inactivates alpha1-antitrypsin which is the primary inhibitor of leukocyte elastase, which is important for leukocyte migration.[129] Normally in the healthy CNS there is a low expression of MMP9 in astrocytes and microglia but its expression increases transiently between 1-2 days post-SCI.[129,129] MMP9 has been implicated in abnormal vascular permeability that is associated with the hemorrhagic injury and inflammation found after SCI.[129] Abnormal increases in MMP9 in both inflammatory cells and endothelial cells can impair the blood-spinal-cord barrier function by degrading the vascular membranes.[11,129]
MMP9 & Neuropathic Pain
Neuropathic pain affects a large percentage of individuals with SCI, and MMP9 has been studied in peripheral nerve injury models of neuropathic pain.[130] After a peripheral nerve injury model, levels of MMP9 rapidly elevate in axons in the dorsal horn of the spinal cord as well as the dorsal root ganglion, where sensory afferents are located.[130] After the peripheral nerve injury, the Schwann cells that myelinate peripheral nervous system axons release MMP9, which is coupled with a pro-inflammatory cascade release of cytokines and growth factors.[130] These damaged and demyelinated axons now experience increased excitability through sodium channels activation, so action potentials continue to be generated even once a stimulus has stopped; thus neuropathic pain is induced and exacerbated by MMP9.[130]
MMP2
Studies on the impact of SCI on MMPs have shown that there is an early increase in MMP3,9,10 and a delayed increase in MMP2,12,13 up through 5 days post-injury.[130] Additionally, there is an upregulation of MMP-2 that correlates with the formation of the glial scar in a contusion model rat model of SCI.[130] A study looking at MMP2-knockout mice and WT controls found there was decreased white matter sparing, decreased nerve fiber regeneration, and an increase in astro-glial scarring with MMP2-inhibition.[130] Thus, interventions to upregulate MMP2 after SCI may lead to improved functional recovery by regulating glial scar formation and white matter sparing.[130]
Clinical Manifestations
The clinical manifestations of spinal cord injury vary depending on the location (cervical, thoracic, or lumbar lesion) and severity of the injury (complete versus incomplete SCI).[2,7]. A list of common clinical manifestations is included in Table 3:
Table 3: Clinical Manifestations of SCI[2,78]
| Body Function | Effects |
|---|---|
| Motor Function | Muscle paralysis below the lesion, spasticity may occur (more common with incomplete SCI), flaccidity can occur with damage to the lower nerve roots below L2, muscle weakness for intact muscles |
| Sensation | Loss of sensation below the lesion, numbness, loss of somatic reflexes. |
| Thermoregulation | Inability to shiver and vasoconstrict blood vessels below the lesion, inability to sweat and vasodilate below the lesion, and excessive sweating above the lesion |
| Cardiovascular Function | Normally the sympathetic and parasympathetic nervous systems work together to adjust blood pressure, heart rate, and distribution of blood flow. Following SCI, there is disruption of SNS input to the heart so the normal balance is lost. This loss in SNS control leads to dilation of peripheral vessels below the lesion, leading to hypotension and bradycardia. There is also decreased venous return of blood to the heart due to decreased tone in the peripheral vessels, loss of muscle pump and changes in intra-abdominal pressure. Decreased venous return leads to decreased stroke volume and cardiac output. |
| Breathing and Coughing | Injuries at or above the T12 level lead to some paralysis of the intercostal muscles, decreased bronchial dilatation, decreased vital capacity, increased risk for pulmonary infections |
| Bowel and Bladder Function | Injuries to the cervical & thoracic cord lead to the development of reflexive bladder/bowel control while lumbar & sacral injuries result in the loss of voluntary bladder/bowel control. |
| Genital Function | Men may experience loss of erections, loss of psychogenic erections, and retrograde ejaculation. Women typically experience loss of lubrication but fertility is not affected. |
Table 4: Secondary Complications of SCI[2,7,9,78]
| Complication | Cause |
|---|---|
| Pressure Ulcers | People with SCI are at risk for pressure ulceres due to motor and sensory impairments as well as skin and circulation changes that make the skin more susceptible to damage. |
| Respiratory Complications | Respiratory complications are a common cause of death following SCI because impaired pulmonary and thoracic muscle function causes reduced ventilation capacity. Ineffective coughing can enable secretions to build in the lungs and lead to pneumonia and respiratory insufficiency. |
| Contractures | Immobilization of joints due to paralysis can lead to contractures and joint deformities. |
| Heterotopic Ossification | Following SCI, bone can form in abnormal locations below the lesion and occurs in as many as 50% of individuals with SCI (males are more likely to be affected). The exact cause is unknown; commonly affected areas include hips, knees, and ankles. |
| Osteoporosis and Fractures | Decreases in bone mineral density, especially of the lumbar spine, femur, and proximal tibia, occur leading to increased risk for osteoporosis and fractures. |
| Pain | Individuals living with SCI commonly experience musculoskeletal pain above the lesion, visceral pain, and neuropathic pain below the lesion. |
| Gastrointestinal Complications | Stress ulcers are commonly developed following SCI due to shock, emotional stress, increased levels of catecholamines, steroid therapy, and increased gastrin production. |
| Urinary Tract Complications | Abnormal bladder function can lead to increased risk for urinary tract infections, kidney and bladder stones, and kidney failure. |
| Deep Vein Thrombosis (DVT)/Pulmonary Embolism (PE) | Peripheral vasodilation, absent or decreased lower extremity muscle pumping, immobility, hypercoagulability, increased blood viscosity, and endothelial damage can lead to an increased risk for DVT/PE. |
| Autonomic Dysreflexia | Autonomic dysreflexia is the excessive activation of the sympathetic nervous system in response to noxious stimuli below the lesion; this potentially life-threatening complication is found in individuals with lesions at or above the T6 level. Stimuli such as bladder distention, catheter blockade, bowel impaction, skin irritations, and infections can trigger a rapid onset of the SNS "fight or flight" response. There is a rapid increase in blood pressure due to vasoconstriction below the lesion, coupled with a drop in heart rate. |
| Cardiovascular Disease | Because people with SCI are now living longer due to medical advances, they are at increased risk from developing cardiovascular disease; this is now the most frequent cause of death for individuals surviving more than 30 years post-injury or over the age of 60. This is likely due to the sedentary lifestyle and increased proportion of body fat commonly seen in individuals with SCI. |
| Orthostatic Hypotension | Orthostatic hypotension is another common secondary impairment which results from an extreme drop in heart rate and blood pressure upon sitting or standing upright. |
Animal Models
Animal models are used to assess biological responses as well to test potential therapies both pharmacological and physical. By using animal models researchers are able to to test different therapies and get a deep look into the changes that are made. The goal for new therapies is to be able to treat the injury as opposed to just treating its consequences and animal model allow researchers to experiment with many different possibilities. The advantages of animal models include the ability to obtain results of different techniques with less concern for safety of the patient, the ability to invasive techniques to investigate the effects of treatments, and the ability to have a repeatable injury so that effects can be seen without having different involvements. Also, animal models allow researchers to control the severity and type of injury. By using animal models researchers theoretically should be able to find the most effective parameters of therapeutic interventions through trial and error. The vast majority of the current models for SCI research is done with mature mice and rats. Other of the more popular types of models include cats, dogs, and some primates. The type of model a researcher uses depends on cost, type of injury to be studied, as well as the hypothesis to be tested. When exploring the neuroprotective strategies of SCI, researchers typically use a contusion or compression injury. when looking at the growth or regeneration of axons researchers often use a transection technique. When transecting the subject researchers can either cut of all tracts (complete) at a particular level or only a few tracts (eg, dorsal column for sensory axons ascending in the fasciculi gracilis and cuneatus). By performing complete transections scientist are able to distinguish if there is any regeneration of axons across the lesion site. Using a in-complete transection, which is the more common type of human injury, allows researchers to look at collateral sprouting and other forms of compensation.[79] Upper SCI injuries are typically used to assess function of the forelimbs in animal models which can be compared to the UE in human counter parts. Lower spinal injuries are used to help evaluate therapies and determine effects of injury for the hindlimbs which compare to human lower extremities.
One of the most widely used and clinically relevant animal models is the contusion injury model in rats. This controlled injury requires exposing the spinal cord by removal of the bone which is usually performed by a dorsal laminectomy. [80] Once the spinal cord is exposed it is then be compressed or transected. The method of the contusion model in rats was originally done using a weight drop technique. For this method, once the spinal cord was exposed a weight was dropped onto the exposed spinal cord producing a repeatable injury. More recently the contusion model is generated using a more controllable device. A displacement driven model was developed at Ohio State University. This device uses an electromagnetic to make a contusive injury to an visible spinal cord. A force driven device, like the Infinite Horizon Device, is also being used in research. [120] These devices allow researchers to produced graded magnitudes of injury as a result of being able to define the amount of force. The devices also allow researchers to produce a reliable and repeatable injury to the exposed rodent spinal cord. The contusion that comes from the device results in an injury to both the gray and white matter of the spinal cord. The area of the cord with the greatest extent of damage is often referred to as the “Lesion Epicenter.” [119] The extent of the injury depends on the amount of force applied.
The transection model involves lacerating a visible spinal cord. As mentioned above transections can be complete or partial. Partial transections are done in order to target to specific areas of the spinal cord (ventral/dorsal/lateral), and cause little additional injury. Complete transection of the spinal cord gives researchers the ability to look at specific levels of injury, their effects, and the possibility of re-generation across a lesion site.[81] Complete transections are severe injuries that are rarely seen in human SCI. In most human SCIs some tissue is spared across the lesion site. Therefore, Incomplete injuries are most often looked at.
Outcome tools
In order to adequately measure the resulting outcomes of such injuries on rats a gross behavioral outcome measure needed to be developed. This measure that is most often used in current research is the BBB (Basso, Beattie, Bresnahan) locomotor scale. This behavioral outcome is used to assess over ground locomotion and has been found to be both sensitive and reliable when measuring overall locomotor performance. [121]. Rating range from 0 -21 and distinguish locomotor features such as flaccid paralysis, isolated hind limb joint movements, and fine details of locomotion. Mice are also being widely used for SCI models. However, because mice have some inherent differences compared to rats the BBB outcome that was being commonly used needed to be replaced because it may have not been accurately reflecting their unique recovery pattern demonstrated by the mice especially when using genetically engineered mice that are extensively used to examine molecular responses to SCI. Therefore, a new outcome measured named the BMS [120] for locomotion was introduced. This outcome model was found to be sensitive, valid, and reliable locomotor measure in SCI mice. The BMS is a 10 point scale (0-9) that uses operational definitions to quantify the magnitude and rate recovery of hind limb movement, forelimb-hind limb coordination, and trunk stability in SCI mice. [120]
Behavioral Recovery in a rat model
Spinal cord contusion was induced in rats with creating 3 different groups based on tissue sparing at the lesion epicenter. The groups included mild (>40%), moderate (15%-30%), and severe (1%-14%). These groups were then evaluated in a sequence of experiments. Locomotor recovery was found to be extensive after mild SCI but limited after severe. In order to find out if the behavioral recovery was due to segmental systems, the spinal cord of rats that showed behavioral recovery with moderate to severe SCIs was transected. By doing this the researchers were trying to find out if the behavioral recovery had been due to the segmental systems (Lumbar spine) only. The study found that transection of the remaining axons eliminated the behavioral recovery resulting in a complete loss of locomotion. Thus proposing that the sparing of tissue at epicenter was responsible for functional recovery. However, after 2 weeks that rats that originally had a SCI and then a transection demonstrated some motor recovery compared to rats that were only transected. This finding showed that when rats recover from even severe SCI, they have altered organizations of the systems below the lesion site.[121] In another animal study involving neonatal opossums [122], the researchers found that opossums were able to restructure their lumbar circuitry during growth in order to allow them to locomote without difficulty following a SCI at 5 days postnatal. Later into adult hood the same opossums were re-transected. Therefore, back limb movements after the transection had to be facilitated due to only the lumbar spinal cord. It was found that the opossums that had their spinal cords transected again showed a substantial regain of hind limb function over time. Both studies showed evidence that restructuring of the lumbar circuits takes place in the presence a few ramaining descending systems. Therefore, by using animals models like the ones mentioned above we are able to tailor interventions and therapies aimed at recovery and not just compensation.
Primate Animal model
Primate animal models is necessarily limited compared with rodent studies. Primate models are much more comparable to human and therefore more weight is put on the results of primate studies. One of the most important factors is the size of the CNS in primates compared to other animal models. In a recent study, 15 adult rhesus primates were studied. The monkeys were given C7 lateral hemi-sections. This model was used to provide a repeatable and measurable deficits in hand function. By using primates, researchers are able to investigate treatments and therapies that may be more related to human SCI. [123] Future SCI animal models would benefit from the use of more primate models, however due to the difficulty of getting primate studies approved rodent models will provide the basis of animal models.
Axonal Regeneration
Axonal regeneration in the adult spinal cord is extremely limited due to many factors that are both intrinsic and extrinsic to the neuron.[82] First, the formation of a glial scar from reactive astrocytosis, which cleans up debris and stabilized the injured zone, hinders axonal regrowth.[3,82,83] Second, at the site of the injury there are various inhibitory molecules present that can be divided into two groups: myelin-associated inhibitory molecules and molecules synthesized by the cellular components of the glial scar, which are present either on the cell or in the extracellular matrix.[3,82,84,85] Myelin-associated inhibitory molecules are the primary axon regrowth inhibitors in the acute phase of spinal cord injury.[3] Types of molecules identified include MAG (myelin-associated glycoprotein), OMgp (oligodendrocyte-myelin glycoprotein), and nogo.[3,82,84,85] There are two types of inhibitory molecules synthesized by the glial scar.[3] One group which includes chondroitin sulphate proteoglycans (CSPG) is solely inhibitory.[3] The other group which includes netrin, semaphorin, and tenascins have both inhibitory and axonal regeneration effects.[3] Lastly, in CNS injury sites neither growth factors nor permissive extracellular matrices are produced to promote regeneration like they are in the PNS.[82] Because axonal regeneration is hampered by various reasons, plasticity of the CNS is responsible for any neurological recovery.
Myelin-associated Inhibitory Molecules
The mechanism by which myelin-associated inhibitory molecules hinder axonal regeneration is related to the collapse of growth cones. Growth cones form when the proximal end of axons are cut and resealed.[3] Growth cones are structures that once elongated form new axons.[3] Centrally, they contain microtubules, mitochondria, and myosin.[3] Actin projections called filopodia have receptors that determine whether or not the axon should extend.[3] Growth cones have receptors on them including Nogo-66 receptor (NgR), low affinity neurotrophin receptor (p75NTR), gangliosides (GD1a and GT1b), and RhoA.[3] NgR binds to all three myelin-associated inhibitory molecules.[3] This binding sets off a cascade that ultimately leads to the collapse of the growth cone.[3] After binding to one of the myelin-associated inhibitory molecules, NgR links with p75NTR, which causes the dissociation of GDI alpha from RhoA and activiates RhoA.[3] RhoA activation is mediated by guanine triphsphate exchange factors (GEF), which adds guanosine triphosphate (GTP) to RhoA.[3] RhoA then binds to serine-threonin kinase (ROK), which activates ROK kinase.[3] ROK phosphorylates the regulatory light chain of the major cytoplasmic myosin, myosin II, and increases myosin II’s activity of actin activated ATPase.[3] ATPase hydrolyzes myosin ATP and begins the contraction between the myosin in the growth cone and the actin in the filopodia.[3] The growth cone collapses because the actin is pulled toward the center of the growth cone instead of out.[3]
Blocking myelin-associated inhibitory molecules activities have been a major strategy in improving functional recovery after SCI. NgR antagonist peptide NEP1-40 (nogo extracellular peptide, residues 1-40) and function blocking fragments have been used to promote both sprouting and regeneration with some functional recovery when administered acutely.[86] Further studies should be conducted to determine if these benefits can still be achieved up to 7 days post injury.[86] Another potential problem is that NEP1-40 does not block MAG or OMgp binding to NgR.[86] This problem resulted in the development of NgR(310), which blocks all three myelin-associated inhibitory molecules and promoted corticospinal tract sprouting and improved behavioral recovery; however, it was administered at the time of injury.[86] Strategies to block intracellular targets such as ROK have also been used.[3] In addition to the pharmacological approach, researchers have also studied the regenerative capacities of NgR-deficient mutant mice.[86,3] Two studies found no regeneration of the corticospinal tract or behavioral improvements in two different NgR-deficient mutant mouse lines.[86,3] The conflicting data between the pharmacological and loss of NgR studies create the need for further research.
Molecules synthesized by cellular components of the glial scar
These molecules have inhibitory and regenerative effects on axons. The members of the CSPG group include NG2, neurocan, versican, brevican, and phosphocan are inhibitory.[3] The mechanism by which these molecules inhibit neurite outgrowth is by binding to growth-promoting lamanin thus preventing it from interacting with its receptor in the growth cone integrin.[3] The members of the group that have both inhibitory and regenerative effects include netrin, semaphorin, and tenascins.[3] Tenascins inhibit neurite outgrowth, but also can bind CSPGs preventing them from performing their function.[3] Netrin and semaphoring are known to contribute to axonal guidance during development.[3]
Plasticity
Plasticity is the capacity of neurons to rearrange their structure and function to adjust to normal development or injury or disease.[87] There are two types of plasticity which are spontaneous plasticity and activity-dependent plasticity. Activity-dependent plasticity will be discussed in detail on the SCI exercise page.
There are two types of spontaneous plasticity which are anatomical and functional. Anatomic plasticity occurs through axonal sprouting in spared intersegmental interneurons, synapse creation, and synaptic rearrangements rostral and caudal to the lesion, and functional plasticity occurs through changes in neuron excitability and inhibition, conduction velocity and synaptic efficiency.[87] Evidence of these changes has existed for quite some time and recent studies confirm them.[82,87,88] Sprouting of spared axons has been observed in both the corticospinal and reticulospinal tracts.[82,89] A study conducted by Ballermann et al.[89] investigated the plasticity of the reticulospinal tract. They knew that the sprouting of spared axons occurred in the corticospinal tract.[89] They also knew that the initiation of walking is governed by the reticulospinal tract and that rats spontaneously recover stepping in weight-bearing in the hind limb on the same side as the injury.[89] This led them to see if the spared reticulospinal tract sprouts like the corticospinal tract.[89] Rats received a lateral thoracic hemisection of the spinal cord.[89] The number of collaterals of spared reticulospinal fibers was compared between recovery after 7 days and 42 days when weight-bearing stepping was recovered.[89] They found that the number of collaterals of spared reticulospinal fibers was directly related with improved locomotor function.[89] Synaptic rearrangement has also been examined in a recent study by Courtine et al.[90] They explored the effects of different combinations of lateral hemisections on kinematics, physiology, and anatomy.[90] The hemisections were different in where they were and when they were delivered.[90] They found that propriospinal projections undergo synaptic rearrangement bypassing the lesion that enhances recovery of locomotion.[90]
Spontaneous plasticity is believed to be responsible for improvements in neurological return, respiratory function, and sexual function.[82,91] Spontaneous plasticity may also be responsible for negative effects such as increased muscle tone, autonomic dysreflexia, and pain.[82,87] The relationship between sensory sprouting and the degree of autonomic dysreflexia was investigated by Cameron et al.[92] They found that sprouting of calcitonin gene-related peptide (CGRP) primary afferent fibers, which is increased by overexpression of nerve growth factor (NGF), results in increased autonomic dysreflexia.[92] Additionally, they found that inhibition of CGRP primary afferent fibers sprouting by overexpression of Semaphorin 3A decreases autonomic dysreflexia.[92] Thus, spontaneous plasticity is not always beneficial and can be detrimental.[87]
Neurotrophic Growth Factors
BDNF
Brain Derived neurotrophic factor (BDNF) is one of many neurotrophins that is involved in synaptic plasticity, neural differentiation, and growth and maturation of both healthy and damaged cells in the neuronal system.[93] The way that BDNF is signaled and expressed in our bodies starts with the TrkB receptor, which is part of the tyrosine kinase family. BDNF is transduced when it interactions with TrkB forming a ligand bond.[93] Then, since TrkB is a tyrosine kinase, it is phosphorylated by BDNF when the ligand bond is achieved, within the intracellular space of the TrkB molecule.[93] From here a variety of cascade events can occur leading to varied expressions of BDNF. There are three pathways which have been recognized in the literature. These include the MAPK, PI3k, and PLC y pathways. From here synaptic proteins can be manufactured, as well as transcription and translation of genes that are involved in axon support and regeneration.[93] The PI3K path has been associated with synaptic plasticity and could be implicated in how the body tries to create trophic support in the face of SCI.[93] In this cascade, the BDNF-TrkB complex leads to activation of the PI3K path when the specific TrkB receptors are utilized by a molecule called Grb2 or otherwise known as growth factor receptor-bound protein.[93] From here, changes in the phospholipid membrane occur allowing protein kinase B to enter.[93] Once activated in the cell, this protein kinase is free to translate genes that help with cell survival and protein trophic support. It has also been proposed that an increase in calcium inside the cell from excited neurons creates activation of BDNF through the PSD-95/TrkB complex.[93] Once this occurs the post-synaptic density protein, hence( PSD-95) can aide axons, dendrites, and therefore quality of synapses so that information is able to be transmitted from cell to cell is a more efficient manner.[93] In the spinal cord injury exercise page, it will be shown that our bodies, with a given stimuli such as exercise, can potentiate these powerful effects that are contained in BDNF and its signaling cascade counterparts.
GDNF
Glial cell line-derived neurotrophic factor (GDNF) is a neurotrophic factor capable of helping damaged axons regenerate after SCI and can also modify and strengthen neural synapses to help promote recovery of function after SCI.[48,128,98,99,100,101] Neurotrophic factors like GDNF tend to respond best when paired with selective receptors. GDNF has been shown to have enhanced expression when coupled with receptors like GDNF-family receptor alpha 1 (GFRα-1) and receptor tyrosine kinase (RET).[48,99,100] These receptors are expressed in neurons, Schwann cells, and glial cells including astrocytes.[98,100] GDNF has also been identified as a player in long-term potentiation (LTP), which is an adaptive plasticity strategy of neurons after SCI and can be involved with chronic pain.[101] Pedersen et. al[101] investigated this role more specifically because many individuals after SCI experience forms of neuropathic pain such as hyperalgesia (increased response to painful stimulus) or allodynia (response to non-painful stimulus).[7,9,101] This study used sciatic nerve stimulation in rats to induce LTP and then studied how it related to the expression of GDNF.[101] Six hours after the induction of LTP, GDNF mRNA expression increased in the dorsal horn of the spinal cord in rats receiving high stimulation frequency-induced LTP.[101]
A study by Keeler et. al[48] looked at changes in expression of GDNF and its receptors GFRα1 and RET in the spinal cord 10 and 31 days after a complete transection.[48] GDNF mRNA and protein levels increased throughout the whole cord by day 10 and began to level decline slightly by day 31 as compared to intact controls.[48] Specifically, the levels increased more in the motoneuron pools of the spinal cord (decline by day 31) but showed a decrease in the intermediate gray matter and dorsal root ganglion by day 31.[48] The receptor GFRα1 increased in the motorneurons by Day 10 and had declined by Day 31.[48] In contrast, GFRα1 showed a steady decline in the intermediate gray matter as compared to the intact controls.[48] In the DRG, GFRα1 decreased initially but began to rise back to control levels by Day 31.[48] The receptor RET levels in the motorneurons increased initially but began to decline by Day 31. In the intermediate gray matter, RET levels did not differ from the controls.[48] In the DRG, RET had a delayed response and showed a slight decrease by Day 31.[48]
A study by Dolbeare and Houle[99] looked at the effects of administering exogenous GDNF after a cervical SCI lesion in rats.[99] Rats were randomized into 4 groups to receive either saline sham injections (2 groups) or GDNF injections (2 groups) and were studied at two different time points (either 1 or 4 weeks post-SCI).[99] The authors found that treatment with exogenous GDNF led to decreased axonal retraction in both the rubrospinal and reticulospinal tracts; these results were significant compared to the groups treated with sham injections.[99] Additionally, one month after the SCI and treatment with GDNF, there were 7 times more regenerating axons than in the control group.[99] This study shows GDNF beneficial after SCI for decreasing the amount of axon retraction as well as improving axon regeneration in two descending tracts.[99] This provides an area where research can be done to maximize the effects of GDNF after SCI, and the effects of exercise on GDNF will be further described on the SCI Exercise page.
NT-3 & NT-4
Neurotrophin 3 (NT-3) and neurotrophin 4 (NT-4) are related neurotrophins involved with regulating synaptic transmission, and help promote plasticity and regeneration in both the spinal cord and muscles following SCI.[102,103,104,127,128] NT-3 and its main receptor, TrkC, have also been found to have a role with helping sensory neurons to survive after SCI.[103] Similarly, NT-4 and its receptor, TrkB, have also been linked to neuronal survival.[127,128] This survival of sensory neurons has even been correlated to improvements in gait pattern.[102,103] Both neurotrophins have been shown to increase with exercise in both the intact and impaired spinal cord, as well as in muscle fibers.[104,127,128] Interestingly, significant increases in NT-4 have been found within spinal white matter, possibly connecting it with oligodendrocytes.[127] In a rat hemisection model of SCI, there was no significant increase in NT-3 mRNA or protein after injury.[103] However, when an exercise protocol was initiated 1 day after injury, there was an increase in NT-3 after 28 days of exercise.[103] This suggests that therapies designed to increase the levels of NT-3 after SCI may be able to promote survival and regeneration of neurons after SCI.[103]
NT-3 is currently being studied for its role in modifying forms of chronic pain such as hyperalgesia (increased response to painful stimulus) and allodynia (response to non-painful stimulus), both of which are commonly found after SCI.[7,9,105] A study by Sharma et. al[105] that NT-3 mRNA and protein levels increase mice muscle tissue subjected to induced-hyperalgesia.[105]
An additional functions of NT-3 and NT-4 are their roles in regulating oligodendrocytes (OL) and their precursor cells (OPC).[106,107,108,127,128] Because oligodendrocytes express the receptors TrkB and TrkC, researchers began investigating whether or not NT-3 or NT-4 could help the proliferation, differentiation, and survival of both OLs and OPCs, which is important because of the vast demyelination and cell damage that occurs after SCI.[3,106,107,108,127,128] If activation of TrkB and TrkC receptors on OLs and OPCs can help produce more OLs as well as aid in remyelination, then therapies that increase these neurotrophins binding to their specific receptors could prove beneficial after SCI.
Medical Management
Introduction
During the secondary mechanisms following SCI, oligodendrocytes die at various stages in response to apoptosis.[94] The implication of this results in demyelination of intact axons which leads to interruptions in conduction leading to further functional deficits. Locomotor recovery to a certain extent can be achieved with as few as 5-10% of myelinated axons in individual tracts remaining.[94] Therefore, a large basis of medical and pharmacological treatment is aimed at preserving and increasing the number of mature oligodendrocytes.[94]
Wallerian degeneration is responsible for the loss of oligodendrocytes which are a distance away from the central lesion.[95] This is compounded by the fact that axonal positive trophic support is decreased at these sites leading to less chance of repairing the damaged cells.[96] Oligodendrocyte apoptosis is thought to result by the activation of cell death receptors such as TNFr and p75NTR. [109] The myelin debris that remains after cell death creates a hazardous environment which has been shown to be inhibitory to axonal re-growth. Many of the treatment approaches detailed below target one or more of these secondary injury pathways.[94] The treatments of Methylprednisone, Stem Cell Transplantation, and Leukemia Inhibiting Factor will be discussed in detail.
Methylprednisone
Not only does the initial injury of SCI cause a hazardous enviornment for cells to thrive, but the secondary injury or damage which occurs only hours after the initial insult sets the stage for many of the functional deficts that are apparent with SCI. A common anti-inflammatory given to patients suffering from an acute spinal cord injury is methylprednisolone. This drug became widely utilized in the early 1990’s after a study in the New England Journal of Medicine showed that when given within 8 hours of injury, neurologic recovery was improved. [110]
Corticosteroids work to reduce inflammation, which has been shown to be responsible for cell death following spinal cord injury.[111] Glucocorticoids are the name given to the body's natural inflammatory reducing cells. The basic process is that glucocorticoids bind to the glucocorticoid receptors which forms a stable complex. From here anti-inflammatory proteins are transcribed and these are allowed to cross into the cell's nucleus where the anti-inflammatory properties are regulated. This is important because the complex actually allows changes to occur via transcription into the DNA. As a result, glucocorticoids, target inflammation via the interaction of various genes and in the case of SCI try to inhibit the negative effects associated with inflammatory molecules. Exogenous Methylprednisone works pretty much the same way that our own natural defenses do. Some benefits associated with this action include staying ahead of the inflammation and reducing the duration and amount that is present following SCI while also preventing our white cells from destroying possibly beneficial substances near the injury site.[112] At low doses, it would be difficult to account for all of the inflammatory mediators at the site of injury so that is why very high doses of methylprednisone are administered following SCI. Validated dose parameters are 30 mg per kg of body weight bolus within 8 hours of injury followed by 5.4 mg per kg of body weight every hour for 23 hours.[110]
Methylprednisone may also combat lipid peroxidation which leads to a host of problems including cell wall degradation via reactive oxygen species (ROS) which take electrons from the phospholipid bylayers rendering them damaged and more prone to apoptosis. Negative side effects of methylprednisone include GI bleeding, pneumonia, myopathy, blood clots, and even interference with neurotrophins.[113]
Figure 11: Lipid Peroxidation

In a study by Lee et al. they showed that methylprednisone preferentially protected the oligodendrocyte immune protecting response, however when looking at neurons and their function, no immunoprotection was observed with Methylprednisone [113] The authors of this study concluded that Methylprednisone may be able to protect the myeling producing cells, however this does not transfer to intact neuron number or quality and may be one reason why current studies show limited effectiveness of the drug. The potential for benefit seems to be intact however in practice MP does not seem to protect the life of neurons at or near the injury site.[113]
Stem Cell Transplantation
Stem cell therapy has been implemented in research models of SCI in hopes of initiating axonal sprouting within the descending motor tracts and in turn, promote functional recovery.[114] The basic premise to this type of therapy is to try and bridge the gap by promoting spinal neurons to form new synapses below the site of injury. Neural stem cells which are transplanted into the injured spinal cord differentiate into glia and neurons.[114] The glial cells help create a good environment for regeneration to occur while the transplanted neurons form the synapse needed to connect with surviving neurons below the lesion, which in the best case scenario leads to improved transmission, and function of the person. Combination therapy has been widely utilized in the literature. Here, chondroitinase, a bacterial enzyme, is used to destroy the inhibitory extracellular matrix component chondroitin sulfate proteoglycan.[115] This is followed by administrtation of neurotrophic growth factors and neural stem cells. This has been shown to act in a positive manner in the repair of chronically injured rat spinal cord.[115]
In a study by Abematsu et al. the researchers epigenetically differentiated neural stem cells into neurons. This is done when valproic acid is used to to basically inhibit the normal structure of the cells in order for differentiation to take place.[114] They then showed that transplantation of these cells led to very good functional recovery in mice following SCI in which the mice could bear weight and walk efficiently which they were unable to do so prior to the treatment. Interestingly no recovery of the corticospinal tract was observed, however new neural synapses sprouting from the graft cells were evident.[114]
Another target of stem cell research is the use of human embryonic stem cell (hESC)-derived oligodendrocyte progenitor cells (OPCs).[116] In addition to differentiating into myelin producing cells, this type of approach also has the proposed benefits of expressing other neurotrophic factors, for example, insulin-like growth factor 1, brain-derived neurotrophic factor, NT-3, nerve growth factor, and transforming growth factor-b1. [116] This leads researchers to believe that not only are the OPCs able to assist in remyelination, but may also be neuroprotective, anti-inflammatory, and promote homeostatic maintenance.[116] In a study by Sharp et al. OPC transplants decreased the severity of the lesion and improved recovery of forelimb function. [116] At the cellular level, both gray and white matter cells were spared to some extent at the the injury site and the largest amount of sparing took place in the motor neurons which are in turn responsible for motor recovery.[116] This research was conducted on a contusion mouse model at the C5 cervical level, and was administered 7 days post contusion injury.[116]
Some of the downsides of stem cell transplantation are that most of the research has been conducted on mice and it is unclear how the results can be interpreted in human analogs. The primary difference in the mouse models is that the specific organization of the motor system compared to humans is different to a certain degree.[114] Specifically, the skills of walking and most voluntary actions and movements are tied more strongly to the corticospinal tract in humans as compared to rodents.[114] It is still unclear if these functional recoveries can also occur to the same extent in spinal cord injuries in humans. A target of further research lies in the ability to differentiate stem cells to mimic other functions of important motor tracts.
Leukemia Inhibiting Factor (LIF)
Leukemia Inhibiting Factor (LIF) has been implicated as a way to lessen apoptosis and help preserve oligodendrocyte survival following spinal cord injury. LIF is a neuroregulatory cytokine which has effects ranging from stem cell differentiation, haematopoiesis, and bone homeostasis.[3] The primary mechanism of action occurs when LIF binds to gp130 via the LIFR-b receptor. This leads to the gp130/LIFR-b complex which is the result of primarily homophilic interactions.[3] The close association of each of the two subunits within this complex leads to cross phosphorylation of the Janus kinases (JAKs).[3] Upon activation, STAT is released by JAK. From here the STAT-3 pathway is attributed to helping oligodendrocytes to survive via anti-apoptotic cells which include Bcl-2, and Bcl-xL.[3] The prevention of apoptosis via the Bcl-2 family is very important in preventing myelin producing cells degradation and ultimate death.[3] Various mechanisms have been proposed on the exact mechanism in which LIF works to promote oligodendrocyte survival, however most sources agree that the protein, suppressor-of-cytokine-signalling (SOCS), can protect fibroblasts from apoptosis induced by TNF-a.[3] So it is possible that LIF, via SOCS, may increase the overall strength of the oligodendrocytes in response to the large accumulation of pro-inflammatory cytokines which are released in response to SCI.[3]
Much about what we know concerning the pharmacological administration of LIF following SCI, has been shown during Multiple Sclerosis research by Butzkueven and colleagues.[117] They utilized an experimental autoimmune encephalomyelitis (EAE) model to further delve into the interactions which showed that LIF can in fact prevent oligodendrocyte death. What they found included an attenuation in oligodendrocyte death with exongenous LIF administration, decreased apoptosis activity specifically occurring via the JAK/STAT pathway, and probably most importantly improved symptomology in the rats being studied. The dose most commonly supported dose in the literature is a daily dose of 25 Kg/kg of body weight.[117]
LIF also has been shown to be neuroprotective in its ability to increase the oligodendrocytes chance of survival when placed in vitro in a cell damaging environment. This is thought to occur either by TNF a, or growth factor withdrawal when LIF is present.[3]
In a study by Azari et. al. it was shown that exogenous LIF given to wild type mice following a hemisection of the spinal cord ellicits some of the same beneficial effects highlighted above. For example the reduction of apoptosis of the oligodendrocytes was found but specially this was mediated through the JAK/ STAT pathway once again, and in addition they also showed that the Akt signaling pathway was implicated as well. This is just another protein kinase mechanism which was shown in this case to express anti-apoptotic mediators.[94] In addition to the Bcl family the cIAP-2 anti-apoptosis molecule was represented and implicated in the improvements.[94] As a result, the surviving axons showed decreased demyelinization as evidenced by preservation of myelin surrounding the axons which where thought to still have the chance of surviving.[94] At the 2 week point, over 50% of mature oligodendrocytes were reduced on both the injured and uninjured side of the spinal cord in the control mice. This was in contrast with a 30% and a 27% reduction in mature oligodendrocytes on the injured and non-injured sides of the spinal cord respectively when LIF was administered.[94]
Chondroitinase ABC
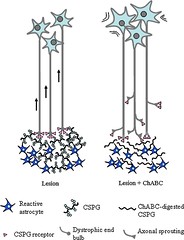
To combat the growth inhibitory regeneration effects of the glial scar that forms after a SCI, recent research has focused on the use of Chondroitinase ABC (ChABC) as a therapeutic treatment both individually and as part of a combination of therapies. Following SCI the body releases glial cells from astrocytes in order to protect the lesion site from further damage. One of the main molecules connected to the glial scar is chondroitin sulphate proteoglycans (CSPGs). These CSPGs are active during development of the CNS and then become a source of stability once the CNS is mature.[124] During SCI, glial cells and CSPGs in particular limit the ability of the body to regenerate axons or to sprout new extensions from existing axons. ChABC theorized to reduce the inhibitory effects of CSPGs by freeing the CS-GAG chains from the CSPG core protein, thus preventing the CSPG from interacting with other molecules in the extracellular matrix and inhibiting neuronal growth. [125] ChABC has been shown both in vivo and in animal studies to increase the ability of the axon to regenerate. In a study conducted by Liu at al.,[126] mature rats with a hemisection were given ChABC intrathecally at different stages after SCI. The authors of the study found that ChABC was able to decrase the amount of astrocytes after SCI which in turn lead to a cellular environment conducive to regeneration. [126]
Overall Summary
An understanding of the timeline of events that occurs post-SCI is crucial to determining the most effective time-frames to provide pharmacologic and exercise-based interventions. Many vascular, immune, and apoptotic factors occur acutely after SCI, while regeneration factors are not necessarily increased without some sort of intervention, such as exercise. An overview of this timeline can be seen in Table 5.
Table 5: Summary of Cellular Components after SCI
| Cell Components | 0-3 Hours | 3-24 Hours | Days 1-3 | Day 3-7 | Beyond 1 Week |
|---|---|---|---|---|---|
| Vascular Changes | Cellular swelling and myelin ruptures within first 15 minutes;[3] Decreased white matter perfusion and hemorrhage in gray matter; White matter perfusion returns to normal.[6] Within 1-3 hours, absent perfusion and disruptions in autoregulation begin; Hemorrhage expands from primary lesion[3], Ischemia progressively worsens[6] | Irreversible necrosis[3] | Reperfusion injury occurs; Hemorrhaging continues[3] | Hemorrhage absent[3] | - |
| NT-3 | - | - | - | May see slight increase but overall exercise mediates any changes in mRNA or protein levels | Again, may see slight change but exercise needed for significant effects[48] |
| BDNF | Ying et al. found a decrease in BDNF mRNA and BDNF expressed protein which occured immediately after injury.[56] | - | A decrease in BDNF mRNA in the lumbar hemicord on the same side as the thoracic lesion by about 15%,[56] however increased BDNF specifically found in astrocytes, microglia, oligodendrocytes, and macrophages as compared to controlled uninjured rats.[57] | Continued increase in BDNF upon immunostaining of microglia, astrocytes, oligodendrocytes, and macrophages.[57] | At six weeks, most of the oligodendrocytes at the injury site tested positive for BDNF, while even 10mm rostral to the lesion, many oligodendrocytes were BDNF positive, which was not the case immediately after injury.[57] In contrast when looking at BDNF mRNA in the lumbar hemicord of a same sided mid thoracic injury, injured rats decreased in percentage by approximately 15% which persisted up to the 28 day marker.[56] |
| NT-4 | - | - | - | - | - |
| GDNF | - | - | - | - | Slight increase in mRNA and protein expression by 10 days post-SCI and values decline by Day 31[48] |
| Immune Cell Markers | Provided there is a direct SCI resulting in immediate compromise of the BBB, some circulating leukocytes may bleed into the site. Microglia are the local immune cells of the CNS and have been found to be active as early as 1 hour following SCI.[58] | Neutrophils first appear attached to venules within 6-12 hours following injury.[3] | Neutrophil concentrations peak.[3] | Hematogenously derived macrophages typically arrive to the lesion site within 2-3 days.[3,59] Neutrophil numbers begin to decline.[3] | Macrophage populations begin declining at 7 days, however remain at the cite in smaller numbers much longer.[60] B-cell populations peak at 7 days and are decreased between one and two weeks post-injury as antigens are removed and endogenous regulatory cascades suppress the response.[61,62] T-lymphocytes persist at the injury long after B-cell numbers decline, up to 1-2 months.[63] |
| Inhibitory Molecules | - | - | At 48 hours post-injury, the glial scar is composed of myelin and oligodendrocyte remnants, which activates the movement of microglia and blood-borne macrophages to the site.[3] During this timeframe, the cellular components of the glial scar except for myelin and its associated inhibitory molecules are permissive to axonal regeneration[3] Phagocytosis of the myelin remnants is believed to be the mechanism of this enhancement.[3] Following the initial events that are permissive to axonal regrowth, the glial scar itself begins to produce its own inhibitory molecules that further prevent axonal regeneration in addition to the myelin-associated inhibitory molecules.[3] | - | - |
| Excitotoxicity | May notice glutamate spikes within first 15 minutes to a few hours. Excitotoxicity spikes anywhere from 1-3 hours | Within 24 hours, glutamate levels have returned to homeostasis | - | - | - |
| Apoptosis and Apoptotic Cellular Components | Within First Hour: Necrosis begins and myelin ruptures.[3] Within 1-3 hours, damage to oligodendrocytes begins and last for weeks[6]; Dysfunction of mitochondria begins[64]; Caspase-3 present in gray matter[43]; | Caspase-3 present in white matter[43] | Apoptosis begins[3]; Caspase-9 found in gray and white matter [43]; Necrotic damage irreversible[3] | 3-7 Days: Apoptosis decreases at primary site but increases rostrally and caudally[3]; Damage to oligodendrocytes peaks[6] | Beyond 1 Week: Apoptosis decreases at primary site but increases rostrally and caudally[3]; Damage to oligodendrocytes peaks[6]; Wallerian degeneration lasts in humans for years[3] |
Go To Spinal Cord Injury Exercise Page
hi
+Intro
++Cell Bio
+++Conclusion
[[f<toc]]
Katie you got a colored block on your picture! Congrats
I figured it out :) Super excited!! I even know how to make the picture larger or smaller!!! "A" for the day!
I figured out tables today! yayyy A for me too!
I figured out hotlinks :)
Awesome - I'm going to clean up the excitotoxicity section and will probably need some hotlinks to connect parts to apoptosis, free radicals, calcium, etc so I'll try to copy your syntax or give you a call later!
Sounds good! I started some overviews for vascular and secondary injury. Feel free to add to them.
I got the bib citation stuff - let me know if you can't figure it out based on my syntax. Once we are completely completely finished, we can go through and change all the name tags to numbers based on order of usage throughout the wiki page.
hey guys just tryin to raise my karma level to two bars!
I need to get one bar of karma…..yikes! I shouldn't have done all of my work on Word!
When I was typing in my references, I had a couple of them that had hyphens in the names. I think because of that a reference number didn't pop up. Does anyone know how to fix that?
Try just using the name before the hyphen within the text and in the actual bibliography.
I really like where you placed your Table of Contents and the pictures. I haven't read through it all yet, but I'll provide more comments when I do.
This page is awesome! I agree with Alyssa…I think the TOC off to the left with the text wrapped looks great! I think you did a great job effectively utilizing pictures and figures to illustrate the text. I love the section on Financial Impact…I think especially for SCI this is important to include. I feel that one could argue the "Cell Bio" of it but I like it :-D I don't have many other constructive comments…because it is very well put together!
hey group Scott showed me how to add links to words so that if you click them it scrolls down to a later part in our page that discusses them. I tried it on free radicals so I'm gonna go through sometime before Thursday and add some more.
I really like this page! The whole section on apoptosis and the components involved is really good. It's easy to understand but also in-depth! You did a really nice job using pictures that are relevant. Great job!